
 Español
Español  Português
Português  English
English Ariel Sánchez
Consiste en la hiperfunción de una o más glándulas paratiroides, con aumento de la concentración sérica de la hormona específica, lo cual provoca hipercalcemia. El hiperparatiroidismo primario es, junto con el cáncer, una de las dos causas más frecuentes de la hipercalcemia (entre ambas entidades, explican más del 90% de las elevaciones patológicas del calcio sérico).
 Figura 75-1 |
El principal agente del sistema homeostático que mantiene el nivel normal de calcio en el líquido extracelular es la glándula paratiroides, cuya función empezó a ser conocida recién en este siglo. La concentración sérica fisiológica del ion calcio es de aproximadamente 1,3 mM; su relación con la secreción de hormona paratiroidea (PTH) es inversa y está graficada en la figura 75-1: al descender la calcemia se produce liberación de la hormona, la cual es inhibida por la hipercalcemia, por ejemplo, la provocada por una infusión intravenosa de calcio, o por la administración oral de un compuesto cálcico de fácil absorción intestinal. La respuesta secretoria ante pequeñísimos descensos de la calcemia es instantánea: ocurre en segundos.
Del calcio circulante total, la mitad está ligado a las proteínas plasmáticas mientras que el resto, o bien forma complejos difusibles (citrato, etc.) o está ionizado. El ion calcio es esencial para la contracción muscular, la excitabilidad neuromuscular, la permeabilidad de la membrana celular y la coagulación de la sangre. Además, el calcio es parte fundamental de la estructura ósea, y sus niveles circulantes deben mantenerse normales para una correcta osificación.
La PTH actúa sobre los osteoclastos para que éstos resorban hueso y aporten al torrente sanguíneo el calcio y el fósforo contenidos en la hidroxiapatita. También actúa sobre el túbulo renal para aumentar la resorción del calcio filtrado por el glomérulo, para favorecer la excreción de fósforo disminuyendo su resorción proximal y distal, y para estimular la síntesis de calcitriol (1,25-dihidroxivitamina D) a partir de su precursor, el calcidiol (25-hidroxivitamina D), por acción de la enzima 1-alfa-hidroxilasa. El calcitriol, a su vez, actúa a nivel intestinal aumentando la absorción de calcio y de fósforo. Cada una de estas respuestas fisiológicas tiende a elevar el calcio total (y/o el iónico) del plasma, mientras que la fosfatemia no varía, o disminuye.
Se conoce bien la secuencia de los 84 aminoácidos que componen la PTH humana, de la cual el primer tercio –el que contiene el extremo amínico– es el que tiene actividad biológica; ya se han sintetizado la PTH1-34 y otros fragmentos de la hormona humana.
En la glándula paratiroides, la síntesis de hormona se hace partiendo de un precursor, la pro-PTH, que tiene 6 aminoácidos adicionales conectados al extremo aminoterminal de la PTH. La pro-PTH es sintetizada en el sistema reticuloendoplásmico rugoso de las células principales, y se trasforma en PTH por medio de una escisión proteolítica en el aparato de Golgi. En glándulas bovinas, el contenido de pro-PTH es de aproximadamente el 10% del de PTH. No hay evidencia de que este precursor sea liberado a la circulación. Hay además un precursor de la pro-PTH, llamado pre-pro-PTH, con 115 aminoácidos (fig. 75-2).
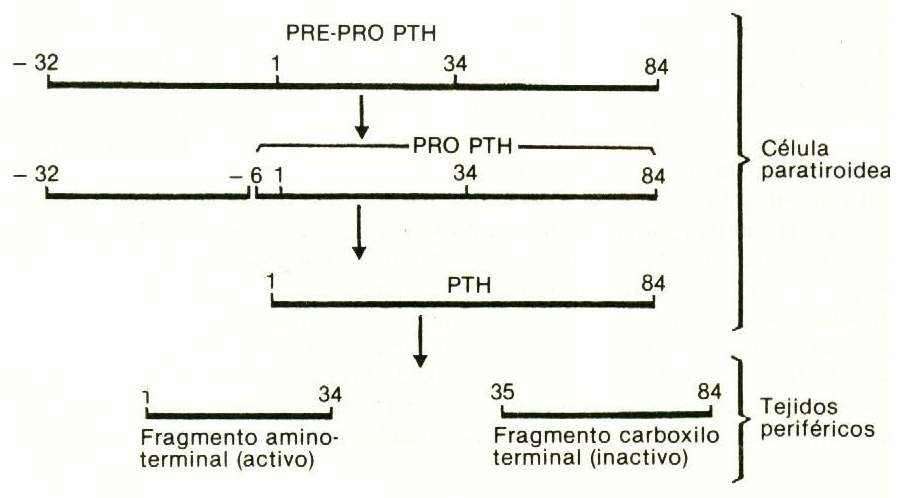 Figura 75-2 |
La hipocalcemia aumenta la conversión intracelular de pro-PTH en PTH. La PTH es almacenada en gránulos secretorios; su liberación depende de que haya una concentración normal de magnesio en el líquido extracelular. Tanto la hiper como la hipomagnesemia la inhiben, quizá por alteración del mecanismo de exocitosis.
En sangre periférica se hallan la hormona intacta o nativa y varios fragmentos, muchos de los cuales no tienen actividad biológica pero sí la identidad inmunológica que los hace reconocibles por los distintos antisueros usados para el radioinmunoanálisis. Esta “heterogeneidad” de la hormona circulante es una de las causas de la dificultad que a veces se encuentra en la interpretación de los resultados informados por el laboratorio. Si bien parte de esos fragmentos es segregada por la propia glándula, la mayoría proviene del metabolismo periférico de la hormona nativa, la cual desaparece de la circulación con una vida media de 1-10 minutos; los sitios principales de esta degradación tisular son el hígado y el riñón. La hormona intacta no es capaz de movilizar calcio del hueso, el cual, por otra parte, no la extrae de la circulación. En cambio, si un hueso aislado es perfundido con PTH 1-34, hay una diferencia arteriovenosa de 36% en el nivel de ese fragmento aminoterminal.
Como todas las hormonas peptídicas, la PTH no penetra en las células para ejercer su acción, sino que se liga a receptores específicos de la membrana celular, los que activan la adenilciclasa y generan monofosfato cíclico de adenosina (AMPc) en el citosol (“segundo mensajero” de la acción hormonal). Cuando esto ocurre en las células del túbulo renal, el AMPc difunde a la luz tubular: sus niveles urinarios aumentan rápidamente después de la inyección intravenosa de la hormona; en realidad, ésta es la respuesta más precoz detectable in vivo.
Fisiopatología
En el hiperparatiroidismo, los mecanismos que deben controlar la secreción normal de PTH están alterados. La secreción es excesiva y autónoma, es decir, no resulta frenada por la hipercalcemia que ella misma provoca. Al contrario de lo que ocurre en otras causas de hipercalcemia, en las que la concentración sérica de PTH es baja, acá coexisten niveles elevados tanto de calcio como de la hormona en plasma.
Basta con que se agrande una de las cuatro glándulas (adenoma) para que se produzca hormona en exceso. Esta hipertrofia uniglandular es la causa más común (80%) de hiperparatiroidismo primario. Le sigue la hiperplasia de las cuatro glándulas, mientras que el carcinoma paratiroideo es poco frecuente (3% de los casos). La pérdida de la sensibilidad normal de las células paratiroideas a los niveles de calcio libre en el líquido extracelular es el principal mecanismo fisiopatológico del trastorno en la mayoría de los adenomas. En la hiperplasia difusa, el set point de las células individuales es el habitual, pero como la masa de células está aumentada, se produce exceso de hormona. No está demasiado clara la base molecular de la enfermedad. El origen clonal de la mayoría de los adenomas paratiroideos sugiere un defecto en el gene que controla el crecimiento de las células paratiroideas o la expresión de la PTH.
La secreción ectópica de PTH –es decir, la producción de la hormona por tumores no paratiroideos–, es un hecho comprobado, pero relativamente poco común. En cambio, es muy frecuente el seudohiperparatiroidismo, que supone la existencia de una sustancia en circulación con efectos muy similares a los de la PTH. La búsqueda de ese misterioso humor llevó al descubrimiento del péptido relacionado con la PTH (PTHrP), que presenta gran homología con la PTH, y que es el responsable de la gran mayoría de las hipercalcemias malignas.
Algunas formas de hiperparatiroidismo son de trasmisión hereditaria. Existen dos tipos de neoplasias o adenomatosis endocrina múltiple (NEM; síndrome de Wermer), que se detallan en la tabla 69-1. En la mayoría de las familias afectadas parece demostrada la herencia autosómica dominante.
Tabla 75-1. Neoplasias endocrinas múltiples
|
NEM tipo 1 |
NEM tipo 2 |
|
Tumores de: |
|
|
Paratiroides |
Paratiroides |
|
Pituitaria |
Tiroides (carcinoma medular) |
|
Páncreas (islotes) |
Médula suprarrenal (feocromocitoma) |
Síntomas y signos
 Manifestaciones osteoarticulares. Se encuentran presentes en la mitad de los pacientes afectados. Suele haber dolores óseos vagos o localizados, y artralgias. Puede darse artritis aguda por depósito de cristales de pirofosfato de calcio bihidratado en meniscos y cartílagos articulares (seudogota por condrocalcinosis), o de cristales de urato monosódico (gota), siendo la hiperuricemia y la hiperuricosuria frecuentes en el hiperparatiroidismo.
Manifestaciones osteoarticulares. Se encuentran presentes en la mitad de los pacientes afectados. Suele haber dolores óseos vagos o localizados, y artralgias. Puede darse artritis aguda por depósito de cristales de pirofosfato de calcio bihidratado en meniscos y cartílagos articulares (seudogota por condrocalcinosis), o de cristales de urato monosódico (gota), siendo la hiperuricemia y la hiperuricosuria frecuentes en el hiperparatiroidismo.
La radiología puede mostrar osteopenia difusa (es el hallazgo más común), aplastamientos vertebrales, quistes óseos en los huesos largos (fig. 76-3), y resorción subperióstica en el aspecto radial de las primeras falanges de los dedos de las manos (fig. 76-4). A veces la calota craneana tiene un aspecto finamente moteado (“en sal y pimienta”) que no es patognomónico. La lámina dura de los alvéolos dentarios puede estar adelgazada o ausente. La calcificación de los cartílagos articulares, el signo típico de condrocalcinosis, solo puede demostrarse radiográficamente; puede haber también calcificaciones periarticulares.
Manifestaciones renales. Un 55% de pacientes tienen nefrolitiasis, de modo que los síntomas y signos clásicos de esta entidad y sus complicaciones (cólico renal, hematuria, infección urinaria) suelen ser la primera forma de presentación de esta endocrinopatía. Los cálculos generalmente son de fosfato o de oxalato de calcio y opacos a los rayos X.
La función renal puede ser normal o hallarse disminuida. Son comunes los trastornos tubulares: pérdida urinaria de bicarbonato, con acidosis sistémica hiperclorémica; disminución de la capacidad de concentración urinaria, con poliuria; glucosuria renal, aminoaciduria, etcétera. La pielografía intravenosa contribuye a evaluar el árbol urinario y además permite descubrir, cuando existen, calcificaciones del parénquima renal (nefrocalcinosis). La ecografía renal es un método no invasivo y útil para evaluar la posibilidad de cálculos.
Manifestaciones musculares. La debilidad muscular y el cansancio fácil son síntomas frecuentes. La primera es más notoria en la musculatura proximal de los miembros: los enfermos tienen dificultad para sacarse una camiseta, ponerse de pie o subir escaleras; puede haber atrofia muscular. La velocidad de conducción nerviosa es normal, pero la electromiografía y la biopsia de músculo muestran alteraciones compatibles con una denervación, sin cambios inflamatorios. Las enzimas séricas (transaminasa glutámico-oxalacética, creatinfosfoquinasa y aldolasa) son siempre normales. No se trata, pues, de una miopatía, sino de una neuropatía.
Manifestaciones gastrointestinales. La úlcera péptica es muy frecuente (más del 20% de los casos). La hipercalcemia produce hipergastrinemia, y ésta lleva a un aumento de la secreción ácida en el estómago. No debe perderse de vista la posible asociación de un adenoma paratiroideo con un gastrinoma (síndrome de Zollinger-Ellison) o con una hiperplasia difusa de los islotes pancreáticos, como parte de una NEM tipo 1. Debe recordarse, sin embargo, que estos casos representan una ínfima parte de los pacientes con la combinación de hiperparatiroidismo primario y enfermedad ulcerosa. La pancreatitis, sobre todo la forma crónica dolorosa, puede ser una complicación digestiva del hiporparatiroidismo. Ya es sabido que la pancreatitis aguda produce una disminución de la calcemia total; por ello, el hallazgo de un nivel normal de calcio sérico en el curso de esa afección debería hacer sospechar la coexistencia de un hiperparatiroidismo.
Otras manifestaciones. Sed, poliuria, constipación, anorexia y molestias abdominales. Es común ver trastornos mentales, con cambios de la personalidad, depresión, y aun psicosis.
Suele asociarse hipertensión arterial, aunque probablemente esto se debe a la alta prevalencia de este trastorno en la población general. La anemia se debe agregar a la lista de complicaciones. Es moderada, normocrómica, normocítica y sin aumento de los reticulocitos (anemia hiporregenerativa). Pueden encontrarse calcificaciones de tejidos blandos como la córnea (queratopatía en banda) y la piel, con necrosis cutáneas.
Metodología de estudio
Si bien el médico debería sospechar un hiperparatiroidismo primario ante cualquiera de los síntomas y signos antes mencionados, debe subrayarse el hecho de que la mayoría de los pacientes son asintomáticos, y que sólo una demostración de su hipercalcemia podría descubrirlos. La hipercalcemia es el único signo invariable y omnipresente en el variado cuadro clínico de esta enfermedad. Sin ella, documentada en más de una ocasión por un laboratorio contable, no se justifica someter al paciente a otros estudios más costosos y que demandan mucho tiempo. El rango normal de la calcemia es de 8,5 a 10,6 mg/dl. Su interpretación exige que se conozca la concentración sérica de las proteínas, ya que una hiperproteinemia puede causar un aumento de la calcemia total, una hipoproteinemia puede enmascarar la hipercalcemia. Debe insistirse en la importancia de realizar varias mediciones del calcio, ya que su elevación puede ser sostenida o intermitente. Esto último ocurre en casos leves, con valores de calcemia en el límite superior del rango normal, o apenas elevados (hiperparatiroidismo “normocalcémico”).
El calcio iónico plasmático está casi siempre elevado. Su medición requiere un aparato especial, provisto de un electrodo de flujo sensible al ion.
Otros signos que deberían buscarse para confirmar el diagnóstico son:
- el aumento de la PTH circulante (la documentación simultánea de hipercalcemia y de un nivel elevado de PTH demuestra la ruptura del sistema homeostático graficado en la figura 69-I); en presencia de hipercalcemia, niveles de PTH en el rango normal alto son compatibles con el diagnóstico. La mayoría de los equipos comerciales existentes en la actualidad miden la hormona “intacta” o “entera” (los 84 aminoácidos) por técnica inmunorradiométrica (IRMA).
- la hipercalciuria (> 400 mg/día);
- la hipofosfatemia (puede estar ausente en pacientes con cierto grado de insuficiencia renal);
- la disminución de la resorción tubular de fósforo (su valor normal debe superar el 85%);
- el aumento de la fosfatasa alcalina –sobre todo de su isoenzima ósea–, que refleja el mayor número y la intensa actividad de los osteoblastos, ocupados en reparar el hueso resorbido por los osteoclastos; está muy aumentada en casos con osteítis fibrosa quística; también aumenta la osteocalcina sérica, otro marcador bioquímico de la función osteoblástica;
- el aumento de los marcadores bioquímicos de la resorción ósea (hidroxiprolina urinaria, uniones de piridinio, péptidos terminales del procolágeno tipo III, etc.);
- el aumento del APMc urinario, que denota el excesivo estímulo tubular por parte de la PTH.
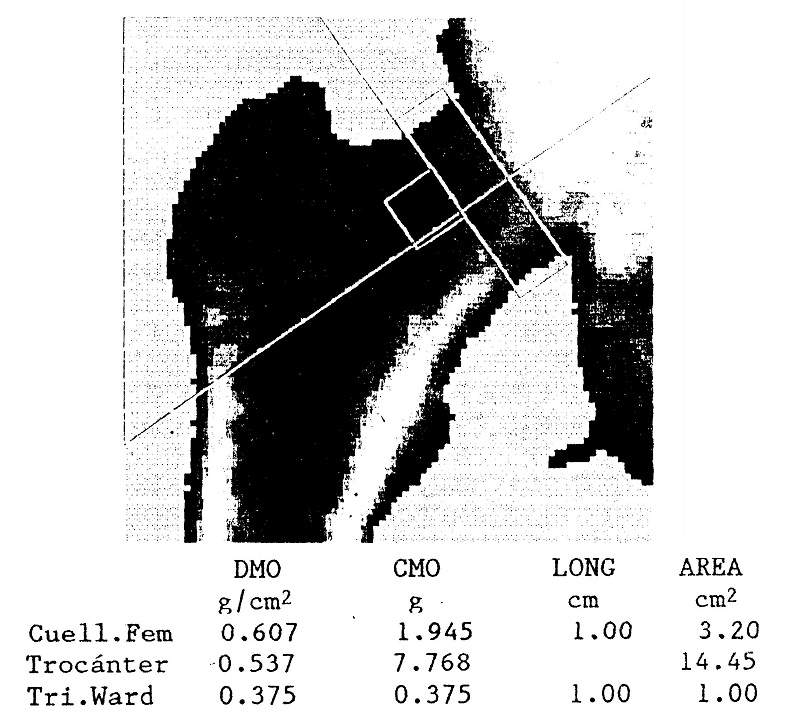
La densitometría resulta mucho más precisa y confiable que la radiografía simple para detectar disminuciones en el contenido mineral óseo. Como el hiperparatiroidismo primario afecta preferentemente el hueso cortical, debe preferirse la evaluación densitométrica de sitios óseos con abundancia de este tipo de hueso, como la diáfisis radial o el cuello femoral.
Es conveniente investigar la calcemia en todo paciente que consulte por:
- úlcera péptica o pancreatitis;
- dolores óseos y articulares crónicos de origen poco claro; osteopenia radiológica; fracturas vertebrales; condrocalcinosis;
- nefrolitiasis cálcica, sobre todo si es recidivante;
- cuadros psiquiátricos mal definidos, con cambios en la personalidad y depresión;
- debilidad muscular proximal en los miembros;
- una combinación cualquiera de las manifestaciones anteriores;
- neoplasias de cualquier origen, con o sin metástasis óseas.