
 Español
Español  Português
Português  English
English Dr. Fernando L. Soldano, Dr. Antonio Molina Rojas y Dr. Gustavo Lavenia
Síndrome caracterizado por hematuria macro o microscópica (signo capital), hipertensión arterial (por retención de H2O y sodio), proteinuria en rango no nefrótico (con o sin edema), y hallazgos en el sedimento urinario, de cilindros hemáticos. Además, puede presentarse clínicamente con insuficiencia renal, y ocasionalmente oliguria. El síndrome nefrítico agudo obedece a varias entidades morfológicas que manifiestan inflamación glomerular difusa. Su presentación puede ser agudo o crónico, en la forma aguda generalmente ocurre tras la infección de las vías aéreas superiores o la piel. En las formas crónicas, es de comienzo insidioso, con alteración progresiva de la función renal, acompañada de proteinuria, hematuria e hipertensión arterial.
Hematuria: indica daño glomerular, puede ser asintomática, micro o macroscópica. La aparición de cilindros hemáticos sugiere inflamación glomerular de cualquier etiología. El hallazgo de, aunque sea 1 cilindro hemático es diagnóstico del síndrome.
Hipertensión Arterial: la alteración del filtrado, retiene sodio y agua, provoca expansión del volumen plasmático, elevando la tensión arterial, como primer mecanismo y también la provoca la activación del sistema renina-angiotensina-aldosterona. La aparición de encefalopatía hipertensiva, suele ocurrir en las formas agudas.
Proteinuria: presente en casi todas las glomerulopatías, aunque en grados variables. Si es leve es asintomática, en los casos severos (> 3.5 gr/24 hs.) se evidenciará clínicamente con edemas.
Edemas: generalizado o localizado (ej. Periorbitario y sacro en la posición decúbito).
Insuficiencia Renal: indicativo de caída severa del filtrado glomerular, aumento de uremia y creatininemia.
Oliguria: Ocurre en algunas formas agudas y es característica de la rápidamente evolutiva, llevando a un síndrome urémico.
En el siguiente esquema, se muestra la evolución que lleva a las manifestaciones clínicas en un paciente con hematuria y disminución del FG de comienzo brusco.
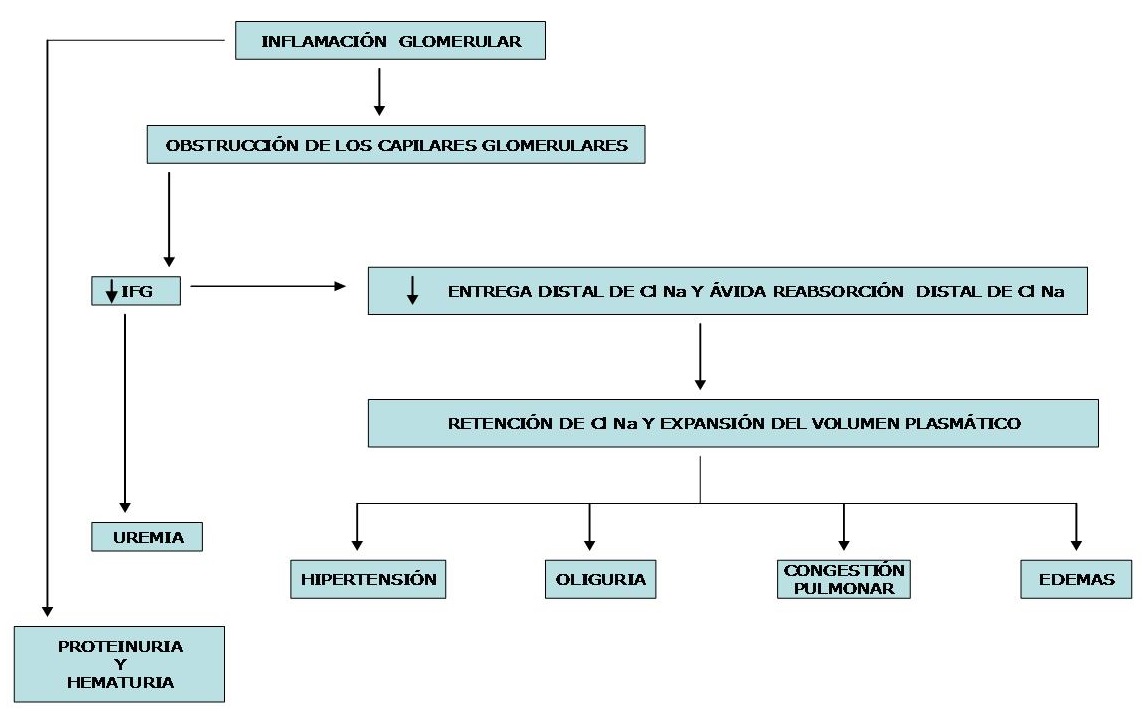
Fisiopatología:
La mayoría de las Glomerulonefritis son mediadas inmunológicamente por:
a) inmunocomplejos (IC): un antígeno que puede ser exógeno o endógeno se une a su anticuerpo en el glomérulo o ambos se depositan en él, previo acoplamiento en la circulación.
b) anticuerpos antimembrana basal glomerular (antiMBG): el anticuerpo que es una IgG endógena, se fija en la membrana basal que es el antígeno.
En ambos tipos de glomerulonefritis la reacción antígeno-anticuerpo activa los componentes del complemento causando quimiotaxis, citólisis y aumento de la permeabilidad vascular. Los neutrófilos liberan enzimas proteolíticas y las plaquetas aminas vasoactivas. Las células mesangiales proliferan y los linfocitos provocan citólisis. La membrana basal tiende a engrosarse, los podocitos se fusionan, se forman trombos y se deposita un material celular acidófilo que oblitera el glomérulo. Esta hialinización (esclerosis) es irreversible. En la flogosis participan prostaglandinas, leucotrienos, serotonina, histamina, angiotensina II, etc.
Las respuestas son variables: si se afectan pocos glomérulos, la GN es focal; si se alteran todos o la mayoría es generalizada. Si los glomérulos afectados están solo en parte, la GN es segmentaria; es difusa cuando lo están en su totalidad. La inmunofluorescencia detecta inmunoglobulinas, componentes del complemento, fibrina, properdina, etc. Cuando estos logran ingresar al espacio de Bowman, estimulan la migración y proliferación de monocitos y células epiteliales formándose semilunas que causan el cuadro de GN Rápidamente Evolutiva.
Se han descripto múltiples modelos experimentales por IC (Inmunocomplejos) que confirman la patogénesis de las GN Humanas. Una es la nefritis de la Enfermedad del Suero: un conejo es inyectado con albúmina bovina y entre 8 a 14 días presenta lesiones articulares, cardíacas y renales. El animal ha desarrollado anticuerpos que con la albúmina bovina forman IC, estos se depositan en el glomérulo produciendo intensa proliferación celular. El conejo sufre HTA, proteinuria, hematuria y oliguria que tiende a resolverse espontáneamente. Las respuestas histológicas y clínicas son variables, con lesiones agudas, crónicas, proliferativas, exudativas, membranosas y esclerosantes. La otra nefritis experimental es la Nefritis de Massugi, en la cual a un conejo se le inyecta un extracto de riñón de rata, produciéndose formación de anticuerpos anti-riñón de rata. Si ahora se inyecta el suero de este conejo a otra rata, esta sufre una GN (fase heteróloga). Steblay demostró que el conejo desarrolla una IgG antimembrana basal glomerular y pulmonar. En una segunda fase, la rata reacciona formando anticuerpos contra la Ig del conejo. Esto provoca un segundo episodio glomerulonefrítico (fase autóloga). En el humano el síndrome de Good–Pasture de etiología desconocida es semejante al modelo de Steblay por su GN y por las lesiones pulmonares. En algunos casos la GN Focal y en otros hay hemorragias pulmonares severas y semilunas que taponan el espacio de Bowman ocasionando oligoanuria.
|
Causas de Síndrome Nefrítico
|
Glomerulonefritis Primarias:
a- Nefropatía mesangial IgA (NIgA):
Denominada así por el hallazgo de IgA como depósito. La NIgA es la glomerulonefritis primaria más frecuente en casi todo el mundo, pero con algunas diferencias geográficas. Las campañas de detección de enfermedades renales y la política de biopsias renales explican en buena medida estas diferencias epidemiológicas. Por razones desconocidas es muy poco frecuente en la raza negra. En adultos, es la primera causa de enfermedad renal biopsiada. Las Nefropatías por IgA pueden ser primarias y secundarias. En las primarias se encuentra la forma idiopática (Enfermedad de Berger) sin manifestaciones sistémicas y con manifestaciones sistémicas (Púrpura Scholein-Henoch). Dentro de las formas secundarias, puede serlo a enfermedades autoinmunes (LES, Enf. De Reiter, Psoriasis, AR, Wegener, Crioglobulinemia Mixta, etc.), enfermedades infecciosas (HIV, TBC, CMV y Toxoplasmosis), y enfermedades neoplásicas (L. no-Hodgkin, mieloma).
Clínica
Es más frecuente en varones, que constituyen alrededor del 74% de todos los casos. El 80% de los casos son diagnosticados entre los 15 y 65 años de edad (adultos). En la Anatomía Patológica, a la MO presenta aumento de matriz mesangial e hipercelularidad mesangial.
Las formas clínicas más comunes son las siguientes: alteraciones urinarias asintomáticas: Es decir, microhematuria asociada a proteinuria no mayor a 1 gr/24 hs. También hematuria macroscópica, síndrome nefrótico y raramente insuficiencia renal aguda.
Diagnóstico
El diagnóstico debe sospecharse ante los cuadros más típicos (alteraciones urinarias o hematuria aislada o recidivante), con estudio urológico normal (radiografías, ecografía renal, y cistoscopia), en ausencia de infección urinaria.
b- Glomerulonefritis proliferativa Mesangial:
Se caracteriza por presentar en la anatomía Patológica, a la Microscopía óptica aumento de la matriz mesangial e hipercelularidad mesangial con pared capilar normal y depósitos densos mesangiales variables. Se clasifican de acuerdo a la Inmunofluorescencia según el predominio de depósitos, en GN por IgM, GN por IgG, GN por C3, GN por C1q, o pueden no hallarse depósitos y solo proliferación. Pueden adoptar formas focales o difusas y existen variedades primarias y secundarias (LES, AR). Las formas clínicas más comunes son las siguientes: alteraciones urinarias asintomáticas: es decir, microhematuria asociada a proteinuria no mayor a 1 gr/24 hs. También hematuria macroscópica, síndrome nefrótico y raramente insuficiencia renal aguda.
c- Glomerulonefritis mesangiocapilar (Membranoproliferativa):
Entre todas las glomerulonefritis, la mesangiocapilar es una de las menos frecuentes. Clásicamente la glomerulonefritis mesangiocapilar (GNMC) se ha clasificado en idiopática y secundaria, siendo subdividida la primera en los tipos 1, 2 y 3 según la histología. Recibe diferentes nombres, también llamada, membranoproliferativa, hipocomplementémica o lobular.
Anatomía patológica:
La característica común de los distintos tipos de GNMC es el aumento de matriz mesangial, la proliferación de células mesangiales y el engrosamiento de la pared capilar.
A continuación, describimos los hallazgos histológicos más típicos de los tres tipos de glomerulonefritis:
GNMC Tipo 1:
Glomerulonefritis mesangiocapilar con depósitos subendoteliales o clásica
Microscopia óptica:
La característica histológica más común es la hipercelularidad endocapilar y el engrosamiento difuso de la pared capilar. Dicha hipercelularidad es debida tanto a la presencia de células mesangiales como de leucocitos principalmente neutrófilos y, en menor medida, eosinófilos. El engrosamiento de la pared se debe a la interposición de matriz mesangial y polimorfonucleares, que confiere a la pared aspecto de doble contorno. Además, hay una disminución de la luz capilar con presencia de "trombos hialinos", que no son verdaderos trombos sino agregados de inmunocomplejos.
GNMC focal
Es una variante de la tipo 1, donde las alteraciones antes mencionadas aparecen sólo en algunos glomérulos y su pronóstico es relativamente favorable.
GNMC Tipo 2
Glomerulonefritis mesangiocapilar con depósitos en la membrana basal o enfermedad por depósitos densos
Microscopia óptica:
Igual que en la tipo 1 se observa proliferación mesangial, aumento de la matriz mesangial y engrosamiento de la membrana basal, pero en un grado más variable, pudiendo simular otros tipos de glomerulonefritis, de ahí que la microscopia electrónica y la inmunofluorescencia desempeñen un papel primordial en su diagnóstico. La lesión típica es una alteración estructural de la membrana basal a expensas de un material densoelectrónico de composición desconocida no siempre visible en la óptica, por lo que para diagnosticar esta nefritis se requiere la observación en microscopia electrónica.
GNMC Tipo 3:
Glomerulonefritis mesangiocapilar mixta (membranosa y proliferativa)
Es una variante de la tipo 1, en la que se observan cambios en la pared capilar similares a los de la membranosa. La inmunofluorescencia muestra depósitos granulares de C3, IgG e IgM, sobre todo, en la pared capilar.
Clínica
La GNMC puede presentarse con cualquier síndrome clínico de enfermedad glomerular, siendo lo más frecuente anomalías urinarias asintomáticas (proteinuria no nefrótica y microhematuria) y síndrome nefrótico que suponen un 30 y 50%, respectivamente. La GN mesangiocapilar afecta a todos los grupos de edad, aunque es más frecuente entre los 5-30 años, siendo excepcional a partir de la séptima década de la vida. Según el tipo histológico hay unas características clínicas distintivas: la tipo 1 se asocia a infecciones con hipocomplementemia y una edad media de debut de alrededor de 24 años. La tipo 2 afecta a pacientes más jóvenes con mayor incidencia de fracaso renal agudo y nefritis rápidamente progresiva. La tipo 3 frecuentemente es asintomática, siendo más frecuente el hallazgo de microhematuria y proteinuria.
d- Glomerulonefritis Extracapilares:
Las glomerulonefritis con proliferación extracapilar, se caracterizan por la existencia de un acúmulo de células en forma de "semilunas" ("crescents", en inglés) que desplazan y ocupan las estructuras normales del ovillo glomerular. Esta proliferación está compuesta por células de aspecto epitelial, situadas en la zona central del ovillo glomerular, motivo que justifica el nombre de proliferación extracapilar. Estas semilunas están formadas por varios tipos de células, entre las que destacan los macrófagos, que han pasado al espacio de Bowman a través de la pared capilar glomerular rota, y las células epiteliales de la capa parietal de la cápsula de Bowman. La lesión inicial que desencadena la formación de semilunas radica en la rotura de la membrana basal glomerular. Esta rotura permite el paso de fibrina y de monocitos al espacio de Bowman. En síntesis, la secuencia patogénica sería la siguiente: 1) depósito de anticuerpos, complejos inmunes y complemento que lesionan la pared capilar, en un proceso en que las células mononucleares y la inmunidad celular parecen desempeñar un papel fundamental, 2) paso de fibronectina y de fibrina al espacio urinario, 3) atracción de monocitos circulantes, 4) proliferación de células epiteliales de la cápsula de Bowman, 5) aumento de la síntesis de proteínas matriciales con formación de semilunas, y 6) evolución hacia la fibrosis, por aumento de la síntesis de colágeno e infiltración de fibroblastos.
Las glomerulonefritis rápidamente progresivas definen, en términos clínicos, un grupo de enfermedades glomerulares caracterizadas por: 1) la presencia de semilunas en más del 50% de los glomérulos, sin contar los completamente esclerosados, y 2) un curso clínico caracterizado por deterioro progresivo y rápido de la función renal, de tal manera que, en ausencia de tratamiento, alrededor del 85% de los pacientes alcanzan la insuficiencia renal terminal en algunos días, semanas o meses. Cuando el porcentaje de semilunas es inferior al 50%, la evolución es más favorable y, en caso contrario, no atribuible a la proliferación extracapilar. Otras entidades que tienen un cuadro clínico similar son: algunas formas de necrosis tubular aguda, nefritis intersticial aguda inmunoalérgica, síndrome urémico hemolítico, enfermedad ateroembólica y riñón de mieloma. Por tanto, la biopsia renal es imprescindible para el diagnóstico de GN EXTRACAPILAR y debe realizarse lo más precozmente posible para evitar retrasos en el tratamiento.
|
Clasificación de las glomerulonefritis extracapilares
|
Clínica
Aparece en pacientes de mediana edad o en edad adulta, y por igual en ambos sexos. El síndrome de Goodpasture suele ser más frecuente en varones jóvenes. En ocasiones hay artralgias, mialgias o un síndrome gripal. Algunos factores ambientales son desencadenantes de todo el cuadro clínico, como tabaco, cocaína, infección respiratoria, edema pulmonar y exposición a hidrocarburos. La afectación renal se caracteriza por oliguria, hematuria, o ambas. La presión arterial es normal o ligeramente elevada. En ocasiones, hay hemorragia alveolar asintomática que se diagnostica tras el hallazgo de macrófagos cargados de hemosiderina en el esputo o por aumento de la captación pulmonar de CO. Como hallazgo de laboratorio, la proteinuria no suele ser nefrótica y la hematuria de origen glomerular es prácticamente constante. El filtrado glomerular disminuye progresivamente con gran rapidez. El complemento sérico es normal. En los casos de hemorragia alveolar aparece anemia ferropénica. El 95% de los pacientes tienen niveles elevados de anticuerpos de tipo IgG α-MBG, detectados con RIA o ELISA. Algunos antígenos del sistema mayor de histocompatibilidad son más frecuentes que en la población sana (HLA-DR2, HLA-DR4 y otros).
Glomerulonefritis Secundarias Postinfecciosas:
Glomerulonefritis endocapilar aguda.
La glomerulonefritis endocapilar se caracteriza por un aumento difuso celular en el ovillo glomerular. La hipercelularidad glomerular se debe a la proliferación, en especial, de células mesangiales, y a infiltración de células inflamatorias. La presentación clínica es variable. La presentación clásica es el síndrome nefrítico agudo, pero, a veces, puede manifestarse solamente con hematuria microscópica y grados variables de proteinuria; más raramente, puede cursar con síndrome nefrótico. Fisiopatológicamente existe un estímulo antigénico endógeno o exógeno y una respuesta inmune que toma como órgano de choque el glomérulo renal. Dependiendo de la etiología, el sistema del complemento se activa. Además, existe una activación local de la cascada de coagulación. Existe infiltración de linfocitos y macrófagos y generación de radicales reactivos de oxígeno que contribuyen a la reacción inflamatoria.
La glomerulonefritis aguda postestreptocócica, es el ejemplo típico de glomerulonefritis endocapilar aguda.
Glomerulonefritis aguda postestreptocócica (GNAPE)
El cuadro clínico se manifiesta de 10 a 15 días posterior a un cuadro de anginas rojas, por estreptococo serotipo M 1, 3, 4, 6, 12, 25 y 49 o posterior a un Pioderma por serotipos M 2, 31, 49, 51, 52, 55, 56, 57, 59, 60 y 61. La lesión es proliferativa y difusa, con abundantes neutrófilos.
Las manifestaciones clínicas son hematuria, edema, hipertensión, oliguria, debilidad, anorexia y, ocasionalmente, dolor sordo en las fosas lumbares. El cuadro clínico completo del síndrome nefrítico agudo (hematuria, edema e hipertensión arterial) se presenta en el 50% de los casos de GNAPE sintomática, pero alguna de estas manifestaciones se produce en el 95% de los casos clínicos. Por eso, los términos glomerulonefritis aguda y síndrome nefrítico se usan a veces en forma intercambiable. Menos de 4% de los pacientes tienen proteinuria en rango nefrótico (> 3,5 g/día) y < 1% tiene una elevación de los productos nitrogenados de curso rápidamente progresivo.
Comúnmente mejora espontáneamente, los pacientes en los cuales persiste proteinuria o recurre la misma, evolucionan a la insuficiencia renal crónica.
Glomerulonefritis Secundarias Multisistémicas:
Glomerulonefritis Hereditaria de Alport:
Es una enfermedad autosómica dominante se asocia con sordera y trastornos oculares. La glomerulonefritis es focal y proliferativa con inflamación intersticial, fibrosis y células espumosas, El mayor porcentaje de aparición es entre los 5 y 20 años y clínicamente se presenta con hematuria, proteinuria, hipertensión y ocasionalmente síndrome nefrótico.
Glomerulonefritis Lúpica:
Se presenta clínicamente con un cuadro de, fatiga, debilidad generalizada, anorexia, pérdida de peso, fiebre, artralgias, eritema malar, dermatosis y alopecia. El glomérulo puede mostrar depósitos de IC con histología normal, proliferativa focal o generalizada, membranosa o rápidamente evolutiva.
Vasculitis Sistémicas:
Constituyen un grupo de entidades clínicos que afectan (inflamación y necrosis de forma preponderante a un tipo y tamaño de vaso y órganos. Entre ellas se encuentran:
- Granulomatosis de Wegener: Causa una GN focal o rápidamente evolutiva, se presenta con afectación del tracto aéreo superior e inferior como epistaxis, rinorrea, sinusitis y deformación del tabique nasal, cavitación pulmonar, tos, disnea y manifestaciones generales (síndrome general): como febrícula, decaimiento general, anorexia y pérdida de peso.
- Panarteritis nodosa (PAN): Se afectan las arterias de mediano y pequeño tamaño, esencialmente a riñan, corazón y sistema nervioso central, se presenta con un síndrome general y suele respetar el pulmón, Afectación cardiaca (70%) en forma de angina, infarto o pericarditis. Afectación renal (80%) con proteinuria, hematuria, cilindros celulares e IR progresiva.
- PAN microscópica: afecta arterias de pequeño calibre, se asocia a G. de Wegener y la glomerulonefritis necrotizante idiomática. Clínicamente se presenta con GN rápidamente evolutiva.
- Glomerulonefritis de Schonlein-Henoch: Es focal o rápidamente evolutiva. Se asocia a púrpura, artritis y cólicos abdominales.
- Enfermedad de Churg-Strauss: granulomatosis alérgica y vasculitis, presenta manifestaciones alérgicas, rinitis, asma y típicamente eosinofilia en sangre periférica. El 70% presenta manifestaciones cutáneas (nódulos subcutáneos en las extremidades, púrpura palpable, rash máculo-papular y petequias). Afectación renal con GN focal y Segmentaria.
Crioglobulinemia mixta esencial:
la GN puede ser proliferativa o membranosa y se puede asociar a púrpura, artralgias y fenómenos de Raynaud.
Anemias Hemolíticas Microangiopáticas: SUH (Síndrome Urémico Hemolítico) y PTT (Púrpura Trombótica Trombocitopénica)
Son entidades diferentes, que comparten mecanismos patogénicos y manifestaciones clínicas comunes. Presentan anemia hemolítica microangiopática, trombocitopenia e IRA. El SUH, es más frecuente en niños, precedida de un episodio de diarrea, entre un 40 a 80 % presentan IRA y 90 % tiene hematuria y proteinuria. La PTT, es más frecuente en adultos, en la tercera y cuarta década de la vida, suele acompañarse además de fiebre y manifestaciones neurológicas.