
 Español
Español  Português
Português  English
English por Juan C. Linares Casas
La cardiopatía isquémica o coronaria es una enfermedad heterogénea que abarca un amplio espectro de manifestaciones clínicas, que van desde el padecimiento asintomático -la isquemia silente- y la angina estable, por un lado, a la angina inestable, el infarto agudo de miocardio, la miocardiopatía isquémica y la muerte súbita, por el otro. Se estima que el 30 al 40% de los eventos coronarios agudos se instalan en personas sin antecedentes clínicos de patología isquémica; por lo tanto, la prevención de los accesos agudos constituye un verdadero desafío en la medicina cardiovascular. Un conocimiento más profundo de sus bases fisiopatológicas puede llevarnos a conformar estrategias de prevención más efectivas.
Fisiopatología.
El sustrato anatómico usual – no excluyente – de la obstrucción coronaria es la reducción de la luz vascular por placas ateromatosas. Estas placas se localizan en las arterias epicárdicas y habrán de generar progresivamente una disminución del aporte de oxígeno por debajo de un nivel crítico de demanda en una zona determinada del ventrículo izquierdo.
1.- Disminución de la oferta de O2: esta oferta está controlada por factores hidráulicos y anatómicos que regulan el flujo coronario:
a) Los factores hidráulicos son: la presión de perfusión coronaria, vinculada a su vez con la presión aórtica; la presión telediastólica ventricular izquierda, y el tiempo de flujo coronario en diástole. Como es sabido, el flujo sistólico al miocardio subepicárdico alcanza al 30% del total, pero el subendocardio no recibe flujo en sístole. Una restricción al flujo coronario en diástole produce así una hipoperfusión marcada del subendocardio.
b) Los factores anatómicos están dados por el lecho vascular coronario y la presencia de ateroesclerosis obstructiva en los grandes vasos epicárdicos. La resistencia al flujo que causan las estenosis ateromatosas cuando superan el 50% de reducción del diámetro vascular genera un gradiente transestenótico con caída de la reserva coronaria, la cual será mayor a medida que la reducción del diámetro se incrementa.
Cabe agregar aquí que una rápida aceleración de las lesiones obstructivas es debida a la instalación de una trombosis sobre la placa ateroesclerosa, pudiendo llegar a la oclusión o suboclusión de la arteria afectada. Muchos autores denominan a este cuadro con el nombre de aterotrombosis, para enfatizar el rol trascendente de este asociación patogénica en los cuadros coronarios agudos.
2.- Aumento de la demanda de O2: la demanda de oxígeno está determinada por la frecuencia cardíaca, la duración de la sístole, la contractilidad miocárdica y la tensión parietal del ventrículo izquierdo.
Todo incremento en alguno de estos factores incrementará los requimientos de oxígeno conducirán a un aumento del gradiente transestenótico y una mayor caída de la reserva coronaria. Es el caso de la estenosis aórtica, la hipertensión arterial y las taquiarritmias.
Sin embargo, los síndromes coronarios agudos y crónicos son procesos activos y no limitados a un ambiente estático de placas ateromatosas fijas. Sus agentes causales son en realidad anormalidades de un entorno molecular y biológico hostil para el endotelio vascular, entorno que es estimulado por un sistema de señales que alteran su integridad, desencadenando reacciones inflamatorias diversas. La placa de ateroma es concebida hoy como el asiento de reacciones bioquímicas entre el endotelio monocelular, la célula muscular lisa subyacente y los elementos sanguíneos que, en otro momento, circulan normalmente. Gracias a la comprensión de estas interacciones celulares y la magnitud de su daño, podemos ahora diferenciar la placa ateromatosa estable de la inestable o vulnerable, sin que esto represente un grado mayor o menor de estenosis arterial coronaria.
La ateroesclerosis es, en síntesis, la consecuencia de una interacción entre diferentes agentes agresivos (dislipemia, tabaco, hipertensión, diabetes) y respuestas defensivas muy complejas. Como resultado de esta “contienda” la pared arterial sufre progresivas alteraciones estructurales y funcionales.
El endotelio normal y patológico.
A pesar de sus dimensiones microscópicas, el endotelio vascular es un órgano endocrino de enorme importancia, cuya integridad es un requisito fundamental para conservar la estructura y la función de la pared vascular, así como la regulación de los fenómenos trombóticos. En un hombre de 70 Kg., tiene una superficie equivalente a 6 courts de tenis, un peso total superior a los dos kilos – más que el hígado – y un número total de células en esa monocapa de un trillón de unidades. Estas células reciben señales y emiten señales. “Sensan” cambios en el flujo, presión, señales inflamatorias o niveles de hormonas o moléculas funcionantes, y responden sintetizando, liberando y activando sustancias que regulan cuatro funciones primarias:
El tono vascular
El crecimiento y proliferación de las células musculares lisas
Trombosis y homeostasis
Respuesta inflamatoria e inmunológica
Como importante órgano metabólico y endócrino, actúa regulando estas funciones de un modo dual: segrega factores relajantes y constrictores vasculares, trombogénicos y antitrombóticos, fibrinolíticos y antifibrinolíticos, inhibidores y estimulantes del crecimiento celular, etc.
Pero debemos destacar que, en condiciones normales, funciona de un modo inhibitorio, facilitando la circulación: segrega factores vasodilatadores, inhibe la contracción del músculo liso vascular, la trombosis, la agregación plaquetaria, el crecimiento celular y la adhesión leucocitaria. Pero en condiciones patológicas, cuando el endotelio se lesiona al ser agredido por diferentes noxas, se “activa” y su función será la contraria: su superficie estimulará la coagulación y la adhesión celular, al tiempo que producirá sustancias vasoconstrictoras y estimulantes del crecimiento celular que engrosarán su pared.
Entre las sustancias vasorelajantes cabe mencionar al óxido nítrico y a la prostaciclina, que son a la vez antiinflamatorios, inhibidores del crecimiento y, en el caso de la prostaciclina, antitrombóticos. Inhiben también el crecimiento celular el factor de crecimiento de transformación beta, la trombospondina y el heparan-sulfato. Como antitrombóticos cabe mencionar a la urokinasa, proteoglicanos de la heparina, el activador tisular del plasminógeno y la trombomodulina.
Esta disfunción endotelial caracteriza a la etapa inicial de la ateroesclerosis, cuando aún no se han producido los cambios estructurales vasculares. Se ha desequilibrado el balance entre los factores endoteliales relajantes y constrictores, con predominio de estos últimos: endotelina, angiotensina II, factor de crecimiento derivado de las plaquetas (PDGF), así como sustancias pro-trombóticas (factor tisular, factor activador de plaquetas, factor de Von Willebrand, inhibidor del activador tisular del plasminógeno), factores de crecimiento vascular y citokinas proinflamatorias.
La producción deficiente o alterada de óxido nítrico (ON) ha demostrado ser la responsable en gran parte de la disfunción endotelial, en respuesta a ciertos estímulos patológicos. Estos estímulos bioquímicos y biofísicos son capaces de desequilibrar la homeostasis del endotelio, dando lugar a un aumento en la permeabilidad de lipoproteínas aterogénicas, al incremento en la adhesión de los monocitos, al estímulo en la producción de citokinas y productos terminales avanzados de reacciones biosintéticas, así como a la alteración en las fuerzas hemodinámicas y biomecánicas que llevan a mayor daño endotelial. Todo ello se traduce en trombosis, inflamación, reactividad vascular anormal, hiperplasia de la intima y fibrosis.
La estría grasa será la primera manifestación histopatológica de la enfermedad ateroesclerosa, que irá progresando hasta constituir la placa fibrosa, que consiste en un núcleo lipídico cubierto por una capa fibrosa que resulta de la síntesis de colágeno, elastina y proteoglicanos por parte de las células musculares lisas y macrófagos que han migrado a la íntima arterial.
El concepto de la Angina de Pecho
El término “angina de pecho” o “angor pectoris” fue introducido por Heberden en 1768 para indicar un “trastorno del pecho” muy característico acompañado de “sensación de estrangulación y ansiedad”. Al emplear la palabra “angina” (estrangulación), Heberden la describía como un tipo de dolor torácico de una modalidad muy especial y que está acompañado de fenómenos psíquicos, el más notable de los cuales consistía en el miedo a la muerte inminente (angor animi).
La angina de pecho constituye un síndrome en sí misma, no una enfermedad. Siempre indica el mismo trastorno fisiopatológico básico: la isquemia miocárdica, sea cual fuere la causa que la genera.
Si bien es casi imposible encontrar una definición clara de la angina de pecho en las publicaciones, se puede establecer que se trata de un síndrome clínico caracterizado por crisis de dolor, opresión o disconfort de origen isquémico, generalmente localizado detrás del esternón, pero ubicado a veces en mandíbula, hombros, espalda o brazos, agravado o desencadenado por el esfuerzo o las emociones, y que se alivia rápidamente al cesar la actividad o el disturbio causal, o por la administración de nitroglicerina.
Algunos elementos arriba descriptos pueden no ser típicos, pero la característica opresiva del dolor o molestia es la base de su reconocimiento. Pueden variar la localización, la irradiación o la duración, e incluso no tener relación con los esfuerzos, pero la sensación opresiva, constrictiva, de pesadez, constituye el carácter más preciso del angor.
Características del dolor.
La localización suele ser retroesternal, si bien a veces el sitio inicial es más amplio e incluye la mayor parte de la región precordial. En ocasiones no se localiza en el tórax: puede afectar la mandíbula, el brazo izquierdo, el epigastrio, los hombros, la región escapular y otras zonas atípicas. En estos casos, su aparición paroxística después de un esfuerzo, su carácter opresivo y su rápida cesación con el reposo permiten sospecharlo.
Si bien puede quedar localizado detrás del esternón, el dolor tiende a irradiar al cuello, al maxilar inferior y a las extremidades superiores. En su forma más clásica irradia hacia la escápula y parte superior del brazo izquierdo. A veces sigue netamente la distribución del nervio cubital a lo largo de la cara anterointerna de manos y dedos.
En cuanto a su carácter, se reitera lo expresado: es un cuadro que clásicamente es constrictivo u opresivo. Se lo ha descripto también como ahogo o ardor. Hay veces en que lo pacientes no refieren dolor sino únicamente opresión o la sensación de un peso sobre el tórax. Puede ser ligero o intenso, pero su calidad es característica. En muchos casos el dolor remeda tanto la distensión detrás del esternón que el paciente intenta vomitar para aliviarse.
Suele obligar al enfermo a mantenerse lo más quieto posible y, si se encontraba marchando, debe detenerse. Se suele acompañar, como ya se mencionó, de una sensación de muerte inminente, que no guarda relación con su intensidad. A medida que las crisis se suceden en el tiempo, los pacientes dejan de sentir esta angustia, por el mayor conocimiento o experiencia que han adquirido ante la enfermedad.
Los episodios de angina suelen iniciarse de forma gradual, alcanzan pronto su máxima intensidad, y desaparecen también de forma paulatina en pocos minutos, generalmente de dos a diez. Se alivian rápidamente con el cese de la actividad que lo provocó, el reposo o la administración de nitroglicerina sublingual. Si el dolor dura treinta minutos o más, debe sospecharse la instalación de un accidente coronario agudo (angina inestable o infarto de miocardio).
Factores desencadenantes.
El esfuerzo es la causa desencadenante más importante y frecuente de la angina de pecho, por incrementar la demanda de oxígeno. El ejercicio físico más común es caminar o subir escaleras. La gravedad del cuadro está en relación con el esfuerzo necesario para generar dolor.
La exposición al frío y la digestión aumentan la posibilidad de que se produzca angor después de un esfuerzo que de ordinario no va seguido de dolor. Asimismo, los estados emocionales pueden desencadenar crisis anginosas a través de mecanismos adrenérgicos complejos.
Pero es importante resaltar que en muchos pacientes los episodios de dolor se producen sin causa aparente y en reposo, como resultado de una caída súbita en la oferta de oxígeno al miocardio, como consecuencia de una ruptura de la placa ateromatosa con trombosis agregada (accidente coronario agudo), o bien como expresión de una obstrucción coronaria de tipo dinámico (espasmo). Más adelante volveremos sobre estos puntos.
La frecuencia con que la angina se presenta en forma atípica y el gran número de enfermedades que se manifiestan con dolor torácico exigen un diagnóstico diferencial cuidadoso, y a menudo difícil. Los dolores punzantes, fugaces y localizados en el precordio suelen ser de origen psíquico. El dolor de la disección aórtica, del neumotórax y, en general, de los procesos que cursan con rotura o laceración de los tejidos, comienza de forma brusca y su intensidad es máxima desde el principio.
La circunstancias y los síntomas que acompañan al dolor son muy útiles en el diagnóstico diferencial. La relación con la ingesta de alimentos o la mejoría con alcalinos orientarán hacia un padecimiento digestivo; los cambios de intensidad con los movimientos de los miembros superiores o el cuello y con la respiración o la posición corporal sugieren una radiculopatía cervical o una pericarditis, respectivamente. Por el contrario, el dolor anginoso, opresivo y angustiante, suele acompañarse de una sensación de gravedad y, en ocasiones, de sudoración y palpitaciones. La disnea durante el dolor no es frecuente, pero su aparición indica una afección coronaria grave y es un signo de mal pronóstico. La presencia de factores de riesgo – hiperlipidemia, diabetes, hipertensión, tabaquismo, antecedentes familiares y un pasado de enfermedad cerebrovascular o de arteriopatía periférica – aumentan la probabilidad de hallarnos antes una patología obstructiva coronaria.
Clasificación clínica de la angina de pecho
Las circunstancias en que aparece el dolor anginoso indican en líneas generales el mecanismo que lo provoca y, a su vez, el conocimiento de éste permite orientar el tratamiento. Atendiendo a estos criterios, se han propuesto diferentes clasificaciones. Consideraremos aquí: a) la angina de pecho estable; b) la angina de pecho inestable, que forma parte de los síndromes coronarios agudos, y c) la angina vasoespástica o de Prinzmetal.
Angina de pecho estable.
El diagnóstico de angina de pecho estable se realiza por la relación entre el dolor coronario y el ejercicio. Se la define como aquella que no modificó sus características en los últimos 3 meses, con o sin antecedentes de infarto de miocardio. Constituye el 60% de los casos diagnosticados.
En general, el nivel de esfuerzo necesario para provocar la angina, o umbral de angina, es constante durante largos períodos de tiempo, de modo que el paciente suele conocer de antemano qué actividades de su vida diaria la provocarán. Cuando un paciente con angina estable cambia las características de su dolor – mayor frecuencia de aparición, esfuerzos provocadores menos intensos, etc.- habrá ingresado en el grupo de las anginas inestables.
Según su severidad y la limitación funcional que impone al paciente, la angina de esfuerzo se divide en cuatro grados siguiendo la clasificación de la Canadian Cardiovascular Society:
Grado I: la actividad física habitual no produce angina, la cual aparece ante esfuerzos importantes, rápidos y/o prolongados.
Grado II: limitación leve de la actividad física; el dolor aparece al caminar con paso normal dos o tres cuadras o subir más de un piso con rapidez, la marcha en pendientes, en el período postprandial, o en climas fríos o contra el viento; en los estados emocionales o al iniciar la actividad matinal.
Grado III: limitación importante de la actividad habitual: el dolor se presenta al subir un piso o al caminar una cuadra con paso normal.
Grado IV: incapacidad para llevar a cabo cualquier actividad física sin la aparición de angina; puede producirse angina en reposo.
Casi el 80% de los pacientes con angina estable pertenecen a las clases I y II. Se ha demostrado que los enfermos en grado III-IV tienen mayor mortalidad que aquellos de los grados I y II.
La exploración física es a menudo normal, especialmente una vez que ha pasado la crisis. Durante el acceso anginoso puede aparecer palidez, diaforesis, taquicardia, así como la auscultación de un cuarto ruido. La presencia de hipotensión y de insuficiencia cardiaca durante las crisis constituyen signos de gravedad.
El electrocardiograma es normal en el 50% de los casos, en ausencia del dolor. El resto puede mostrar signos de un infarto antiguo, hipertrofia ventricular izquierda, depresión del segmento ST o cambios isquémicos en la onda T.
La prueba ergométrica constituye en estos pacientes un test muy importante para el diagnóstico. Se considera positiva si durante la misma, o inmediatamente después, aparece angina o si el segmento ST desciende al menos 1 mm adoptando una forma horizontal. Si el dolor aparece precozmente (menos de 6 minutos) o si la depresión del ST supera los 2 mm o desciende la presión arterial durante el esfuerzo, estaremos ante un paciente de alto riesgo que deberá ser referido a estudio coronariográfico con miras a una revascularización.
Los estudios radioisotópicos de imagen y el ecocardiograma de esfuerzo permiten identificar extensión, severidad y localización de la isquemia. La coronariografía y la tomografía multicorte nos informarán sobre el estado de la función ventricular y la extensión de las lesiones coronarias. Es importante destacar que la sobrevida está en relación con el grado de ateroesclerosis coronaria: a mayor número de coronarias obstruidas y a mayor deterioro contráctil del ventrículo izquierdo, peor será el pronóstico. En tal sentido, los hallazgos de la coronariografía constituyen los mejores factores de predicción.
En cuanto a la morfología coronaria en la angina estable, se debe enfatizar que las imágenes de las placas ateromatosas son las de un ateroma no complicado, esto es, de superficie lisa, blanca amarillenta, sin ulceraciones, hemorragias intimales o trombos adherentes. En esta indemnidad de la cubierta fibrosa de la placa radica la diferencia con las lesiones de tipo inestable.
Angina de pecho inestable.
Algunos tipos de angina de pecho se consideran formas inestables de la enfermedad coronaria y la conducta terapéutica difiere sensiblemente del de la angina estable, por cuanto su presencia indica una situación de evolución imprevisible. Bajo el término de angina inestable podemos incluir los siguientes tipos: a) la angina de comienzo reciente (duración de los síntomas inferior a un mes); b) la angina progresiva: es aquella donde las crisis dolorosas se hacen más frecuentes o de mayor duración, son rebeldes a la nitroglicerina o se presentan con esfuerzos cada vez menos intensos; c) el tercer subgrupo está constituido por la angina de reposo.
La angina inestable integra el llamado grupo de los síndromes coronarios agudos, por lo que será analizada más adelante.
Angina de Prinzmetal.
La angina de Prinzmetal o angina variante constituye una manifestación clínica particular de la cardiopatía isquémica que se caracteriza por crisis dolorosas en el reposo y elevación del segmento ST durante las mismas (a veces en forma de onda monofásica).
Este cuadro responde a un incremento en el tono vasomotor coronario o a un espasmo en el vaso, que puede instalarse sobre una coronaria sana o ateromatosa, y que va a ocasionar isquemia a través de una disminución en la oferta de oxígeno.
El electrocardiograma suele ser normal fuera de las crisis anginosas, especialmente en ausencia de lesiones coronarias ateroesclerosas.
Los pacientes con espasmo coronario presentan una respuesta muy acentuada a la administración de ergonovina, que reproduce el cuadro clínico y electrocardiográfico de la angina variante. Se trata sin embargo de una prueba potencialmente peligrosa que debe evitarse si no se conoce el estado del árbol coronario.
LOS SINDROMES CORONARIOS AGUDOS
Los síndromes coronarios agudos (SCA) constituyen una causa frecuente de consulta en la práctica médica diaria. Su importancia radica en la elevada morbimortalidad y el alto costo sanitario que genera, vinculado esto ultimo al uso de medicación y realización de exámenes paraclínicos y la realización de procedimientos diagnósticos y terapéuticos invasivos.
Definición y clasificación
SCA es un término operacional que ha sido desarrollado para referirse a una constelación de síntomas compatibles con isquemia miocárdica aguda. Esto abarca un espectro variado de entidades clínicas que van desde la angina inestable (AI) hasta el infarto agudo de miocardio (IAM) en todas sus variantes.
Los SCA se clasifican según las alteraciones del segmento ST del electrocardiograma (ECG) inicial en:
- SCA con elevación del segmento ST; y
- SCA sin elevación del segmento ST.
Esto tiene su justificación basada en que el primer grupo de pacientes serán considerados como portadores de un infarto agudo y la estrategia de mayor importancia terapéutica es la reperfusión miocárdica de emergencia mediante trombolisis o angioplastia.
En el segundo grupo se presentan los pacientes con angina inestable e IAM sin elevación del segmento ST, diferenciándose entre si por la ausencia o presencia respectivamente de marcadores biológicos y enzimáticos en sangre; asimismo, el manejo terapéutico inicial es el mismo para ambas entidades.
Fisiopatología de los síndromes coronarios agudos
Los SCA comparten un origen fisiopatológico común (Fig. 1): el accidente de la placa ateroesclerosa, con erosión o ruptura de su cubierta fibrosa y, como consecuencia de diversos tipos de injuria, fuerzas hemodinámicas y probablemente inflamación, expone un sustrato altamente trombogénico que al interactuar con la sangre genera dos hechos sobresalientes a) la activación y agregación plaquetaria y b) la generación de trombina. Ambos hechos interactúan y dan origen a un trombo de mayor o menor magnitud.
Si el trombo es oclusivo generará la interrupción total del flujo coronario en el vaso culpable y usualmente la consecuencia es el infarto de miocardio transmural o infarto-Q (con elevación del segmento ST), mientras que si la trombosis agregada no es oclusiva usualmente el cuadro clínico acompañante es un SIA sin elevación de ST, es decir, una angina inestable o un infarto-no Q.
La placa vulnerable o de alto riesgo.
A pesar de que las lesiones ateromatosas tienen la misma fisiopatogenia, las placas son muy heterogéneas y aquellas “de alto riesgo” tienen características propias. El conocimiento de la enfermedad ateroesclerosa avanzó desde la vieja idea que sostenía que la oclusión aguda de una arteria era la consecuencia final de una obstrucción lentamente progresiva, al concepto actual que establece que es el producto de la rotura de una placa, con la formación posterior de un trombo que obstruye parcialmente u ocluye totalmente el lumen arterial. Pero sólo recientemente se ha postulado que es más importante la composición de la placa como indicador de riesgo de rotura que el grado de obstrucción previo, convirtiéndose en el factor determinante de esta enfermedad.
La placa ateroesclerosa propensa a la rotura, llamada también “vulnerable” o de “alto riesgo” tiene, desde el punto de vista histológico, un gran núcleo lipídico, por lo general excéntrico y con depósitos de lípidos extracelulares, alta densidad de linfocitos-T y macrófagos plenos de lípidos, reducido número de células musculares lisas, y todo ello cubierto por una delgada capa fibrosa (Fig. 1). Su consistencia es la de una pasta dentífrica a temperatura ambiente, siendo aún menos consistente a la temperatura corporal. Por lo tanto, no es para sorprenderse que estas placas sean poco estables y con tendencia a la ruptura, más aún si las comparamos con otras placas fibrosas y ricas en colágeno (Fig. 2).
Una vez rota o erosionada la delgada capa fibrosa, su interior lipídico es expuesto a la corriente sanguínea que desencadena la cascada de la coagulación, generando un trombo que ocluye u obstruye la luz, con las manifestaciones clínicas de los síndromes coronarios agudos (Fig. 3).
 Figura 1
Figura 1
 Figura. 2: La placa estable tiene una capa fibrosa relativamente gruesa que protege al núcleo lipídico del contacto con la sangre.
Figura. 2: La placa estable tiene una capa fibrosa relativamente gruesa que protege al núcleo lipídico del contacto con la sangre.
 Figura 3: Esquematización fisiopatológica de los Síndromes Isquémicos Agudos.
Figura 3: Esquematización fisiopatológica de los Síndromes Isquémicos Agudos.
IAM Q: infarto de miocardio tipo Q o transmural; AI: angina inestable;
IM no Q: infarto agudo de miocardio tipo no Q.
La angina inestable nos sugiere que ha tenido lugar una erosión o fisura relativamente pequeña de una placa vulnerable que da origen a episodios transitorios de oclusión trombótica con la consecuente angina de reposo. Este trombo suele ser lábil, con oclusiones que no exceden los 15 o 20 minutos. Además, la liberación de sustancias vasoactivas por parte de las plaquetas va a originar vasoconstricción secundaria, lo que puede contribuir a reducir aún más el flujo coronario.
En el infarto no-Q el daño de la placa es más importante y resulta en oclusiones trombóticas más persistentes, que pueden durar hasta una hora. Una cuarta parte de los pacientes con infarto no-Q presentan oclusión coronaria por más de una hora, pero el territorio miocárdico isquémico suele estar irrigado por colaterales.
En el infarto tipo Q o transmural ocurre una fractura mayor de la placa que puede resultar en la formación de un trombo fijo y persistente por más de una hora con la consecuente necrosis transmural del miocardio comprometido. Algunos casos de muerte súbita coronaria se asientan probablemente en una lesión rápidamente progresiva en donde la ruptura de la placa y la trombosis resultante conducen a fatales arritmias ventriculares.
Clínica
Los pacientes con angina inestable e infarto de miocardio suelen quejarse de dolor anginoso con características similares a las de la angina estable, pero son más intensos y prolongados. Así como en la angina de esfuerzo clásica el dolor dura entre 2 y 10 minutos, aproximadamente, en la angina inestable persiste entre 10 y 25 minutos, aparece con esfuerzos menos intensos e incluso en el reposo.
En el infarto de miocardio el dolor anginoso suele durar más de 30 minutos – a veces horas- , y a menudo es más intenso. No se alivia con nitroglicerina y puede acompañarse de arritmias o síntomas y signos de insuficiencia cardiaca. No se intensifica al sentarse o al respirar profundamente, criterio útil para diferenciarlo del dolor de la pericarditis aguda.
La mayoría de los pacientes con infarto, especialmente aquellos con supradesnivel del segmento ST, se encuentran angustiados y agitados, y tratan sin éxito de aliviar su dolor moviéndose en la cama, modificando su postura. Es frecuente encontrar palidez junto con sudoración y frialdad de las extremidades. La combinación de dolor torácico retroesternal de más de 30 minutos de duración y sudoración es un sólido argumento a favor de un infarto agudo de miocardio. Además, se suele auscultar un galope presistólico apexiano (R4), debido a la contracción enérgica de la aurícula izquierda a causa del aumento en presión diastólica ventricular izquierda.
La presión arterial y la frecuencia cardiaca pueden mostrar hipertensión y taquicardia o bien hipotensión y bradicardia, según exista hiperactividad simpática o parasimpática, respectivamente, aunque en los infartos con elevación del ST la presión sistólica suele caer unos 10 o 15 mm Hg.
Después de los primeros días de instalado el infarto, existe un frote pericárdico en el 6 al 10% de los casos, que suele ser intermitente y se ausculta con mayor claridad a nivel del ápex o en el borde esternal izquierdo; puede persistir varios días, y se debe a una pericarditis localizada por la extensión transmural de la necrosis al epicardio.
Suele presentarse fiebre después de las 24 horas del infarto, generalmente de grado moderado y que persiste entre dos y cuatro días. A veces existen vómitos, que a veces se deben a la medicación utilizada.
En cerca de la mitad de los casos se detecta un factor desencadenante previo al infarto, en general un ejercicio físico intenso y desusado, un stress emocional o una enfermedad médica o quirúrgica. Puede ocurrir en cualquier momento del día o de la noche, pero su frecuencia alcanza su máximo valor en las primeras horas del despertar. Este pico circadiano se debe a la combinación del incremento del tono simpático y la mayor tendencia a la trombosis – mayor agregabilidad plaquetaria - que ocurre entre las 6 de la mañana y las 12 del mediodía.
En caso de disfunción ventricular izquierda, shock cardiogenico e insuficiencia mitral isquémica, se hallarán los signos propios de tales complicaciones.
Antes de seguir adelante, debemos subrayar un punto que no debe olvidarse: alrededor del 25% de los infartos no se reconocen clínicamente; la mitad de ellos cursan de forma asintomática y el diagnóstico se realiza retrospectivamente al analizar un electrocardiograma. En el resto, el dolor es atípico o no está presente. Algunos autores dan porcentajes aún mayores de infartos “silentes”.
Electrocardiograma.
El electrocardiograma (ECG) es de capital importancia y permite clasificar a los SCA en dos grandes grupos: con y sin elevación del segmento ST. Los primeros desarrollarán por lo común un infarto de miocardio con onda Q (infarto transmural), mientras que los segundos presentan con alta probabilidad una angina inestable o un infarto sin onda Q. La diferencia entre estos dos últimos cuadros estará dada por la presencia o ausencia de marcadores bioquímicos de necrosis en los exámenes de sangre, como se verá más adelante.
Siempre que se afecte un espesor suficiente de la pared ventricular izquierda, y más concretamente en los infartos transmurales, se producen cambios e el ECG debidos a: a) la isquemia; b) daños miocárdicos más avanzados o zona llamada “de injuria”, y c) la necrosis o infarto propiamente dicho. Estas alteraciones se desarrollan dinámicamente y en forma evolutiva, sucesiva, condicionando las siguientes modificaciones:
En un inicio, en los primeros minutos de la oclusión coronaria, la isquemia modifica la repolarización ventricular y produce la inversión de las ondas T, que aparecerán negativas, puntiagudas y profundas en las derivaciones que miran a la zona isquémica. Por tanto, enlos infartos de pared anterior probablemente las T sean negativas en las derivaciones precordiales; en los infartos laterales, lo serán en D1 y aVL; y en los infartos pósteroinferiores la T estará invertida en D2, D3 y aVF.
Al acentuarse la isquemia y el daño miocárdico consecutivos aparecen las llamadas “corrientes de lesión” o de “injuria” en las derivaciones que miran a la lesión, y que se traducen en el ECG por una elevación del segmento ST, elevación que llega a su máxima expresión al 1º o 2º día del evento. Esta sobreelevación del ST puede incluso llegar a ser de tipo onda monofásica (Fig. 4).
El área de necrosis miocárdica no genera potenciales de acción. Si el infarto abarca todo el espesor de la pared ventricular (infarto transmural), el mismo puede ser considerado como una ventana abierta en la pared. Por tanto, el potencial negativo del interior de la cámara ventricular se transmite a través del tejido necrosado a las derivaciones que miran al área infartada. En lugar de la onda positiva normal, el comienzo del complejo QRS ventricular estará dirigido hacia abajo, esto es, habrá ondas Q profundas y anormales, y por complejos electrocardiográficos solamente negativos del tipo QS. Estas ondas Q son más anchas y profundas que las ondas Q que se observan normalmente en algunas derivaciones. Aparecen varias horas después de iniciado el cuadro, sucediendo a los cambios en el segmento ST. Su aparición tardía obliga, por lo tanto, a reparar en los cambios electrocardiográficos más precoces a fin de clasificar a los SCA y proceder a las conductas terapéuticas apropiadas según cada caso.
La onda de lesión o desnivel positivo del segmento ST en, entonces, fundamental para el diagnóstico de infarto transmural y su extensión es proporcional al tamaño o extensión del mismo. Este tejido lesionado aparece desde las primeras horas que siguen a la oclusión y persiste, habitualmente en descenso paulatino, de 3 a 4 semanas, evolucionando hacia la necrosis total (ondas Q profundas) o, al menos en parte, hacia la isquemia simple, indicando en este último caso una mejor evolución. A medida que mejora el supradesnivel del ST va a existir entonces un aumento de la isquemia en el subepicardio (ondas T negativas).
En el caso de los síndromes coronarios agudos sin elevación del segmento ST, esto es, la angina inestable y el infarto no Q.
En la angina inestable el ECG puede no mostrar cambios, pero en la mayor parte de los casos pueden observarse alteraciones transitorias: infradesnivel del segmento ST mayor de 1 mm o mayor de 0,5 mm, inversión de las onda T, o incluso bloqueo de rama izquierda de grado avanzado y arritmias.
En el infarto no-Q sucede que el tejido necrosado no ocupa todo el grosor de la pared ventricular, y por lo tanto la onda de activación puede alcanzar por su camino habitual a la zona transmural sana. No tiene lugar aquí la “ventana” eléctrica de los infartos Q. Podemos observar disminución en el voltaje de las ondas R en las derivaciones que miran a la zona infartada, infradesnivel del segmento ST, ondas T invertidas (Fig. 5) y bloqueos de rama, al igual que en la angina inestable.
La diferencia entre los distintos cuadros coronarios agudos sin elevación del segmento ST no debemos buscarla en el ECG sino en los marcadores bioquímicos de necrosis.
 Figura 4: ECG de un infarto agudo con elevación del segmento ST, evidenciable en las derivaciones II, III y aVF
Figura 4: ECG de un infarto agudo con elevación del segmento ST, evidenciable en las derivaciones II, III y aVF
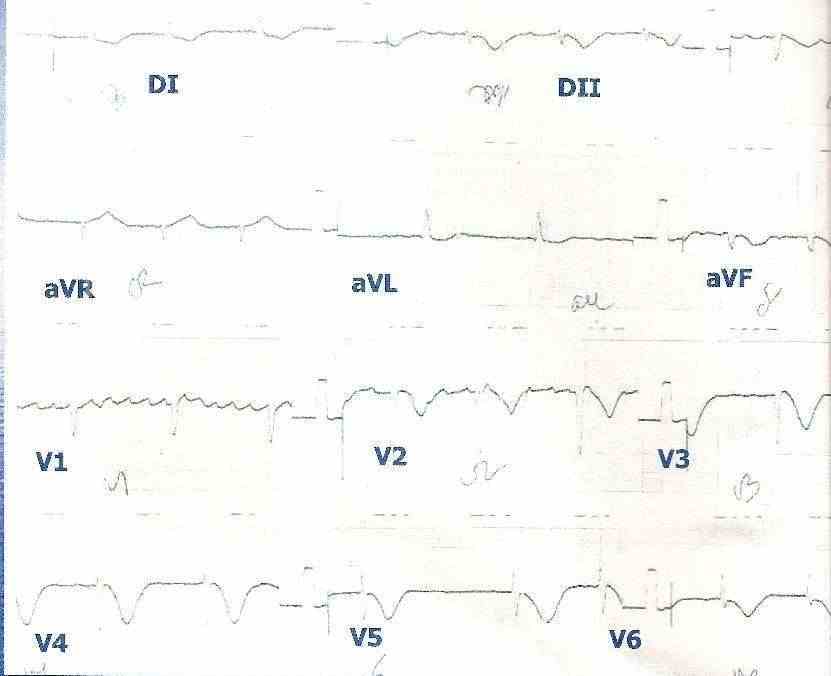 Figura 5: Síndrome coronario agudo sin elevación del segmento ST en un paciente con fibrilación auricular crónica. Se puede observar en las derivaciones V2, V3, V4, V5 y V6 la inversión de las ondas T.
Figura 5: Síndrome coronario agudo sin elevación del segmento ST en un paciente con fibrilación auricular crónica. Se puede observar en las derivaciones V2, V3, V4, V5 y V6 la inversión de las ondas T.
Marcadores bioquímicos de necrosis miocárdica
El infarto de miocárdio ocasiona diversas alteraciones humorales, como leucocitosis y aumento de la velocidad de eritrosedimentación. No obstante, desde el punto de vista diagnóstico solo tiene importancia la aparición en sangre de diferentes proteínas liberadas en el torrente circulatorio, liberadas por los miositos dañados: mioglobina, troponinas cardíacas T e I, creatinfosfokinasa (CPK), lácticodehidrogenasa (LDH) y otras. Se establece el diagnóstico de IAM cuando los niveles hemáticos de biomarcadores específicos y sensibles, tales como la mioglobina, troponinas y fracción MB de la CPK, están incrementadas en el contexto clínico de la isquemia aguda. Estos marcadores reflejan daño miocárdico pero no indican su mecanismo. Por lo tanto, un valor elevado en ausencia de evidencia clínica de isquemia nos indica que se deben pesquisar otras causas de lesión cardiaca, como la miocarditis.
Se utilizan por lo general dos marcadores. Lo más recomendado es usar una combinación de un marcador de ascenso rápido, como la mioglobina, y de otro que demora más en subir pero que es más específico, como las troponinas, a fin de detectar la presencia de infarto tanto en pacientes que se presentan precoz como tardíamente.
La mioglobina es detectable en sangre a los dos o tres horas del inicio. Su concentración asciende rápidamente, alcanza un máximo nivel entre las 6 y 12 horas del comienzo de los síntomas y cae a valores normales en las siguientes 24 horas, eliminada por los riñones.
Las troponinas cardíacas se consideran como los marcadores de uso corriente de mayor especificidad cardiaca en el diagnóstico de la injuria miocárdica, en particular, la troponina I y la troponina T. Estas proteínas se asocian con secuencias específicas de aminoácidos codificadas por genes diferentes a aquellos que codifican las isoformas musculares esqueléticas. Así, la TnI tiene completa especificidad para el miocardio, en tanto que la TnT se presenta en pequeñas cantidades en el músculo esquelético durante el desarrollo fetal humano y es reexpresada en patologías que se vinculan con la regeneración muscular (por ejemplo, la distrofia muscular de Duchenne). Las troponinas aparecen en el suero de modo relativamente precoz tras el inicio del infarto (4 a 10 horas), alcanzan su pico en 48 horas y permanecen anormales durante 4 a 10 días.
Si no se dispone de la determinación de troponinas, la mejor alternativa es la CPK-MB: es menos específica que la troponina cardíaca, pero su especificidad clínica para la injuria irreversible es más robusta. Al igual que con las troponinas, una CPK-MB aumentada (por ejemplo, para el diagnóstico de IAM) es aquella que excede el 99% de los valores de un grupo de referencia.
Las mediciones de la CPK total no suelen ser recomendadas para el diagnóstico de rutina del infarto, en razón de la amplia distribución tisular de esta enzima. Sin embargo, su larga trayectoria hace que muchos médicos aún la utilicen, aunque en estos casos es deseable combinarla con un biomarcador más sensible. Siempre se debe descartar el uso previo de inyecciones intramusculares.
Las transaminasas y la dehidrogenasa láctica y sus isoenzimas no deberían usarse para el diagnóstico del infarto agudo, ya que su ascenso puede acontecer en otras circunstancias clínicas: embolia pulmonar, acidosis, lesiones hepáticas y musculares, así como por la administración de algunos medicamentos como las estatinas. Solo se recurrirá a ellas ante la imposibilidad de contar con marcadores más específicos.
Así, en un síndrome coronario agudo sin elevación del segmento ST, la presencia de marcadores bioquímicos anormales nos hablará de la existencia de una angina inestable, en tanto que una elevación de los mismos confirmará que nos hallamos ante un infarto no-Q.
Ecocardiograma
El ecocardiograma, en la etapa aguda de los eventos coronarios, descubre anormalidades de la motilidad de las paredes en 89% a 100% de los pacientes con infarto trasmural, con una disminución de la sensibilidad entre 79% y 86% 6 para los infartos no trasmurales. Los infartos pequeños pueden no producir alteraciones ecocardiográficas. Existen limitaciones técnicas por malas ventanas acústicas y por la presencia de entidades como bloqueos de la conducción y miocarditis.
Pronostico
Los pacientes se puede clasificar en 3 grupos de riesgo:
a) Riesgo alto. Aquellos con angina de reposo, recurrente o acelerada y que además presentan una de las siguientes características: inestabilidad hemodinámica (hipotensión, insuficiencia cardiaca, disfunción mitral); descenso del ST > 1 mm transitorio o persistente; aparición de nuevo bloqueo rama o arritmias ventriculares; angina postinfarto (en el primer mes); después de revascularización coronaria (en el primer mes); edad > 75 años; troponinas notablemente elevadas (troponina T > 0.1 ng/ml). Este tipo de pacientes tienen un riesgo superior al 5 por ciento de sufrir un infarto de miocardio o morir en los primeros 30 días, por lo que se recomienda la realización de cateterismo cardíaco en las siguientes 24 horas con vistas a revascularización.
b) Riesgo medio. Pacientes con angina inestable más ausencia de características de alto riesgo, inversión de onda T > 2 mm en 2 o más derivaciones y la presencia de algún modificador de riesgo (enfermedad coronaria previa, arteriopatía periférica o enfermedad cerebrovascular conocida, diabetes mellitus, revascularización previa, troponinas > 0.01 ng/ml).
c) Riesgo bajo. El resto de pacientes sin características de riesgo alto o medio, y electrocardiograma normal.
Complicaciones del infarto
Las complicaciones del infarto agudo de miocardio son muy frecuentes. Pueden presentarse en el momento de desencadenarse el episodio o bien desarrollarse posteriormente, pero es en los primeros dias cuando ocurre con mayor frecuencia.
Estas complicaciones son: 1) arritmias y trastornos de la conducción de la conducción auriculoventricular; 2)insuficiencia cardiaca; 3) soc cardiogénico; 4) insuficiencia mitral aguda; 5) comunicación interventricular aguda; 6) ruptura de miocardio; 7)pericarditis y síndrome posinfarto; 8) angina posinfarto; 9) infarto del ventrículo derecho; 10)embolia pulmonar, y 11)embolias sistemáticas.
1.Arritmias. La incidencia de arritmias durante la evolución de un infarto agudo de miocardio varia según los distintos autores entre un 70 y un 100% de los casos. Esto depende de muchos factores (edad, uso de drogas, acidosis, tamaño del infarto, etc) pero fundamentalmente esta dada por los diferentes métodos de detección, ya que cuando se emplea el monitoreo osciloscopico, su frecuencia de aparición es menor que cuando se lo hace con regristo en memoria electrónica o cinta magnética. Siguiendo a Lown, es util dividir a las arritmias en dos tipos: 1)aquellas producidas como resultado del proceso isquemico (falla eléctrica), y 2) las secundarias a una descompensación del estado hemodinámico por severo deterioro miocárdico (falla mecánica).
En ultima instancia, ambas son consecuencia de una alteración electrofisiológica de las miofibrillas, pero en las segundas se deberá encarar, además, la terapéutica de la falla de bomba.
Se describirán brevemente las arritmias sobre la base de su lugar autónomo de origen
a) Taquicardia sinusal. Denota hiperactividad simpática y se le encuentra en el 30% de los casos; es mas frecuente en los infartos anteriores que en los inferiores. Las causas mas comunes son: dolor, angustia, insuficiencia cardiaca, pericarditis e hipovolemia. Las que requieren un preciso diagnostico diferencial son la falla de bomba y la hipovolemia, pues el tratamiento es muy diferente en ambas. Un dato muy util es el registro de la presión capilar pulmonar con catéter de Swan-Ganz: su medida decidirá si existe hipertensión venocapilar (mas de 18 mm Hg) o por el contrario, hipotensión (menos de 19 mm Hg.)
b) Bradicardia sinusal. En el momento inicial del infarto ocurre en el 40% de los casos, pero en la unidad coronaria se la observa en solo el 20%. Es mas frecuente en los infartos inferiores que en los anteriores, en una proporción de 3:1, posiblemente por hiperactividad parasimpatica.
Es una arritmia por inestabilidad eléctrica, en razón de la isquemia nódulo sinusal, reflejos vagales o pericarditis. En ocasiones se asocia a extrasistolia ventricular por aumento del automatismo
c) Extrasistolia auricular. Ocurre en el 20% de los casos y se debería a un aumento del automatismo o a fenómenos de reentrada auricular, secundarios a falla de bomba, hipoxia o trastornos electrolíticos.
d) Fibrilación auricular. Se la observa en el 10 al 15% de los pacientes y puede indicar precozmente insuficiencia ventricular izquierda o infarto auricular. Tambien puede responder a causas extracardiacas, como hipokalemia, hipertiroidismo, hipoxia o embolia pulmonar. Como la sístole auricular contribuye al 15% de la fracción de eyección ventricular, la perdida de las misma en un paciente infartado puede ser fundamental para el mantenimiento de un apropiado gasto cardiaco. Asimismo si la respuesta ventricular es alta (superior a 130 latidos/ minutos) se deteriorara el estado hemodinámico, observándose signos de descompensación cardiaca(oliguria, estertores basales, hipotensión) por acortamiento del periodo de llenado diastólico y por aumento del consumo miocárdico de oxigeno, que a su vez puede extender la necrosis. Esta arritmia pues debe ser tratada de inmediato.
e) Aleteo auricular. Presente en el 1 al 2% de los IAM, al igual que la fibrilación puede deberse a una insuficiencia ventricular izquierda o a un infarto auricular. Según el grado de bloqueo AV, la frecuencia cardiaca oscilara entre 75 y 300 latidos/ minuto. En los infartos inferiores se suelen observar bloqueos 3:1 y 4:1 por isquemia o injuria del nódulo AV. Con estos grados de bloqueo se evidencian bien las típicas ondas del aleteo.
f) Taquicardia auricular paroxistica. Ocurre en el 5% de los pacientes infartados. La frecuencia cardiaca oscila entre 150 y 300 latidos por minuto y su fisiopatología es la misma que en la extrasistolia auricular.
g) Ritmo de la unión AV (nodal). Su frecuencia oscila entre un 2% y un 15% según diferentes autores. Constituye un ritmo de escape, por depresión del nódulo sinusal o bloqueo AV como conse¬cuencia de la isquemia.
h) Bloqueo auriculoventricular (bloqueo AV). El bloqueo AV de primer grado ocurre en el 9% de los casos, el de segundo grado en el 8% y el de tercer grado en el 7% de los pacientes que ingresan en la Unidad Coronaria con un IAM.
De todo el sistema de conducción auriculoventricular, la unión AV es el sector mas sensible a la. anoxia e isquemia. Por ello, en los infartos agudos inferiores es frecuente observar distintos grados de trastornos de conducci6n AV como consecuencia del compromiso de la irrigación normal del nódulo AV.
El bloqueo AV de primer grado (todos los estímulos atraviesan la unión AV, pero lo hacen mas lentamente), o tiempo de conducción AV de mas de 0,20 segundos, no requiere tratamiento pero si un control cuidadoso, pues puede evolucionar a grados mayores de bloqueo; esto ultimo se ve mas frecuentemente en los infartos anteriores.
Los bloqueos AV de segundo grado (no todos los estímulos atraviesan la unión) responden a dos mecanismos básicos. En el tipo Mobitz I (o blo¬queos con periodos de Wenckebach) se observa una prolongación progresiva del segmento PR, lo cual indica una conducción cada vez mas afectada, hasta que una onda P se bloquea totalmente. Esto prolonga el tiempo disponible para que el nódulo AV se recupere, y la onda P siguiente presentara una conducción AV normal, lo cual da origen a un nuevo ciclo de prolongación progresiva del PR.
Este tipo de bloqueo AV es mas frecuente en los infartos inferiores y se debe habitualmente a isquemia del nódulo AV. Aun sin tratamiento puede desaparecer en 72 horas. El bloqueo AV tipo Mobitz I constituye el 90% de los bloqueos AV de segun¬do grado.
El bloqueo AV de segundo grado tipo Mobitz II es menos frecuente y se observa en menos del 10% de todos los pacientes con IAM que padecen un bloqueo. En el mismo se observa la falta de conducción brusca de una onda P sin alargamiento previo del segmento PR. En la mayoría de los casos se asocia a un infarto anterior y se debe casi siempre a la injuria por debajo del haz de His, que a menudo progresa hacia un bloqueo completo. Por ello este trastorno requiere, en general, la implantación de un marcapaso transitorio en el ventrículo derecho.
En el bloqueo AV de tercer grado (completo) las aurículas y los ventrículos laten independientemente.
Se lo observa en pacientes con infartos inferiores o anteriores, y se origina en el edema que rodea al tejido infartado y que compromete al sistema de conducción. En los infartos inferiores la lesión es nodal, con desarrollo gradual del bloqueo y aparición de ritmos de la unión AV de 40 a 60 latidos por minuto. En los infartos anteriores el bloqueo AV se instala bruscamente y es causa de alta mortalidad. Se produce por necrosis extensa del tabique que involucra al haz de His (lesión infranodal) y puede terminar súbitamente en asistolia, falla de bomba o fibrilación ventricular. El bloqueo AV de tercer grado es indicación formal de marcapasos transitorio endocavitario y estimulación a demanda.
i) Trastornos de la conducción intraventricular. Se observan en el 10 al 20% de los casos. Pueden ser: 1) bloqueo completo de rama derecha (BCRD); 2) bloqueo completo de rama izquierda (BCRI); 3) hemibloqueo anterior izquierdo (HAJ); 4) hemibloqueo posterior izquierdo (HPI); 5) BCRD con HAI, y 6) BCRD con HPI.
Los bloqueos completos de rama pueden evolu¬cionar a un bloqueo AV de tercer grado en el curso de un infarto agudo. Los IAM complicados con BCRD o BCRI tienen alta mortalidad hospitalaria.
j) Extrasistolia ventricular. Es la arritmia de observaci6n mas comun en el IAM, con una fre¬cuencia superior al 75% de los casos. Obedece a un desequilibrio eléctrico a nivel celular producido por la isquemia (arritmia por inestabilidad eléctrica). Como por lo general no existe descompensación hemodinámica, no son de mal pronostico, pero deben ser tratadas si aparecen mas de seis por minuto, si son multifocales, bigeminadas o trigeminadas o si con anterioridad han originado arritmias graves.
k) Taquicardia ventricular. Ocurre en el 20% de los pacientes en las primeras horas del infarto agudo. Se la define generalmente como una sucesión de latidos ventriculares (cuatro o mas) con una frecuencia inferior a los 120 latidos / minuto.
Es mas frecuente en los casos de infarto trasmural y en aquellos con severa falla ventricular, y el mecanismo electrofisiológico responde al aumento del automatismo de las fibras de Purkinje lesionadas (foco ectopico) y a fen6menos de reentrada. Es una arritmia grave que requiere rápidas decisiones terapéuticas.
l) Ritmo idioventricular acelerado. Se lo define como un ritmo ventricular con una frecuencia de 60 a 100 latidos por minuto, y ocurre en el 15% de los pacientes con IAM. Se lo observa al segundo o tercer dia del cuadro, y responde a un mecanismo de escape, por enlentecimiento del ritmo de base o bien a un foco ect6pico de taquicardia ventricular. Los episodios suelen ser pasajeros y tienen buen pronostico.
m) Fibrilación ventricular. Esta arritmia grave se manifiesta en el ECG por una sucesi6n de ondas lentas, polimorfas y de aparici6n irregular. Se presenta en el 10% de los casos y es la principal causa de muerte en los pacientes con IAM antes de que ingresen a Unidad Coronaria. Puede ser eléctrica o primaria y mecánica o secundaria. La primera es la que ocurre como un hecho abrupto en un paciente sin datos previos de insuficiencia circulatoria, mientras que la mecánica es la arritmia terminal de un paciente con severa falla de bomba.
2.Insuficiencia cardiaca (falla de bomba). La falla ventricular izquierda en el curso de un IAM continua siendo el desafío mas importante al accionar del medico en la Unidad Coronaria. La pérdida de miocardio funcionante es la causa mas importante de la insuficiencia cardiaca en este cuadro, pero en muchas ocasiones existen otros factores que contribuyen a desencadenar, mantener o agravar la insuficiencia ventricular. Entre ellos se incluyen la disminuci6n de la precarga por hipo-volemia relativa o absoluta, el aumento de la poscarga originado por la descarga adrenérgica en las primeras horas del I AM, la presencia de arritmias y las complicaciones mecánicas posinfarto, tales como la discinesia ventricular, o la ruptura de músculo papilar o del tabique interventricular.
Desde el punto de vista clínico, siguiendo el criterio de Killip y Kimball, se clasifica a los pa-cientes en cuatro clases funcionales:
Clase I: pacientes no complicados, sin manifestaciones de insuficiencia cardiaca.
Clase II: síntomas o signos mínimos de insufi¬ciencia cardiaca (estertores basales, galope o hipertensión venosa).
Clase III: edema agudo de pulmón por severa
falla ventricular izquierda.
Clase IV: shock cardiogenico.
El monitoreo hemodinámico en la Unidad Coronaria, mediante la colocación en el lecho capilar de la arteria pulmonar (que representa las presiones telediastólicas del ventrículo izquierdo) de un catéter de Swan-Ganz de termodilución, es de gran importancia para una correcta evaluación de estos pacientes. Atendiendo al estado de congestión pulmonar y de perfusi6n perif6rica, Forrester y Ganz los clasifican en:
Grupo I: índice cardiaco superior a 2,2 1/min/m2 y presión capilar pulmonar inferior a 18 mm Hg. El paciente esta asintomático.
Grupo II: la presión capilar pulmonar es superior a 18 mm Hg. y el gasto cardiaco se mantiene en cifras superiores a los 2,2 1/min/m2.
El paciente puede estar asintomático o presentar signos clínicos y radiológicos de congestión pulmonar mas o menos severa. Si el cuadro clínico es de edema agudo de pulmón y las resistencias sist6micas estan muy elevadas, puede presentar además signos de vasoconstricción periftiica que simulan una situación de bajo gasto.
Grupo III: la presi6n capilar pulmonar es inferior a 18 mm Hg. y el gasto cardiaco menor de 2,2 1/min/m2. En esta situación, que es de hipovolemia absoluta (por vómitos, diarreas o diuresis excesiva) o relativa (en la cual no existe una aut6ntica hipovolemia, pero debido a la disminución de la contractilidad el ventrículo necesita una precarga mas elevada para mantener una funci6n normal), el pa-ciente puede estar asintomático o pre¬sentar signos de hipoperfusion periférica e hipotensión arterial.
Grupo IV: la presión capilar pulmonar es superior a 18 mm Hg. y el índice cardiaco infe¬rior a 2,21/min/m2. En esta situación de grave deterioro hemodinámico el pa¬ciente presenta prácticamente siempre signos clínicos de congestión pulmonar y de bajo gasto. La presion arterial suele estar reducida. La forma mas grave de insuficiencia cardiaca, el shock car¬diogènico, este vinculada en este grupo. Para hacer el diagnostico de shock cardiogenico es preciso que la presión capilar pulmonar sea alta y descartar asi el shock hipovolemico.
3. Shock cardiogenico. La Myocardial Infarc¬tion Research Unit (M.l.R.U) del National Heart and Lung Institute de Bethesda define asi al shock cardiogenico:
a)Presión arterial sistólica menor de 90 mm Hg., o inferior en 30 mm Hg. a su valor anterior en un hipertenso, durante por lo menos media hora, asociado con:
b)Inadecuada perfusión periférica manifestada por los siguientes criterios clínicos bioquímicos:
1. Debito urinario inferior a 20 ml/hora y natriuria por debajo de 30 mEq/1.
2. Piel fría, pálida, lívida sudorosa y viscosa.
3. Alteración mental (hipoperfusion cerebral) caracterizada por agitación, somnolencia o coma.
4. Compromiso del metabolismo aer6bico celular (acidosis láctica).
La incidencia de shock cardiogenico después de un I AM es del 15% aproximadamente, y la mortalidad es abrumadora, excediendo el 90% en algunas series. Los estudios anatomopatológicos de-mostraron que este cuadro se presenta cuando mas del 40% de la masa miocárdica esta dafiada, ya sea por injuria reciente o asociada a necrosis antigua. La extensión del dafio esta directamente relacionada con el pronostico y es el resultado de una lesión progresiva, constituyendo un circulo vicioso de isquemia, depresión miocárdica y mayor isquemia.
Una necrosis extensa de la masa ventricular ocasiona una reducci6n de la contractilidad, una disminución del gasto cardiaco y un descenso de la presión arterial. El volumen sist61ico decae rápidamente y, si dicha caída alcanza entre el 30 y el 50% de su valor normal, se produce hipoxemia tisular, con el consiguiente metabolismo celular anaer6bico y el desarrollo de acidosis láctica.
A través de la hipoxemia y de la liberación de catecolaminas (mediada por activación simpática) se instala un mecanismo de retroalimentaci6n negativo con un aumento consecutivo de la resistencia vascular fundamentalmente en el riñon, el territorio esplacnico y la piel, de tal modo que el volumen de sangre que retorna al corazón derecho disminuye.
Ello explica que, si bien aumenta discretamente la presión telediastolica ventricular izquierda, en el 75% de los casos no se produce edema pulmonar ni aumento de la presión venosa central.
La hipotensión, la liberación de noradrenalina y la reducci6n del volumen circulante efectivo determinan hipoperfusion renal y hepática. La primera estimula el sistema renina-angiotensina-aldosterona, y la reducción de la perfusión hepática merma el metabolismo de la aldosterona en el hígado. Estos dos mecanismos producen aumento de la aldosteronemia, con la retención consiguiente de sodio y perdida de potasio a nivel renal. La necro¬sis miocárdica, la anoxia, la acidosis y el déficit de potasio son terreno fértil para el desarrollo de arritmias graves.
El mantenimiento de un gasto cardiaco bajo y de la hipoperfusion por varias horas pueden causar un mayor daño miocárdico, estableciéndose una retroalimentación positiva e irreversible entre disminución del gasto cardiaco y daño miocárdico. El mecanismo de este circulo vicioso parece responder a la hipoperfusion del miocardio y a un factor humoral que deprime la contractilidad de la célula miocárdica. La hipoperfusion del miocardio, con la consiguiente disminución de la contractilidad, extiende el área infartada. Entre los factores humorales se destaca la producción de polipéptidos activos que deprimen la fuerza de contracción miocárdica y que se producen principalmente en el páncreas y el territorio esplacnico por la ruptura de lisosomas consecutiva a la hipoxia de estos tejidos.
Asi, el circulo vicioso que nace de las complicaciones del shock, si no se resuelve precozmente conduce a la muerte con rapidez. En lo relativo a las características cómicas de este grave cuadro, se puede agregar que los pacientes se presentan gravemente comprometidos, con síntomas y signos secundarios a la mala perfusión tisular, como por ejemplo: confusi6n mental, intranquilidad, vaso-constricción de tegumentos, cianosis periférica, pulsos poco amplios, hipotensión, taquicardia, oli¬guria (diuresis menor de 20 ml/hora), etc., y a la congesti6n pulmonar: disnea, ortopnea , polipnea, etc. Los indicadores mas sensibles de perfusión tisular son el estado del sistema nervioso central y el flujo urinario, elementos que deben ser evaluados en forma permanente.
Para llegar al diagnostico de shock cardiogenico por IAM, se deben tener en mente los siguientes elementos:
A)dolor precordial prolongado en las ultimas 48 horas;
B)en ocasiones edema agudo de pulmón sin valvulopatia previa, uremia o intoxicación;
C)shock no debido a intoxicación, sepsis o hemorragia;
D)angor rebelde.
Cualquiera de los casos anteriores debe presentar además signos electrocardiográficos de infarto agudo y aumento de las enzimas pericas.
Un paciente que retina los criterios anteriores, y además los que se mencionan en la definición ori¬ginal, se encuentra en shock cardiogenico.
Para cuantificar los trastornos hemodinàmicos, su evoluci6n y el efecto de las medidas terapéuticas, se debe realizar un control con equipos de monitorización hemodinámica. Un instrumento al que ya se hizo referencia, de gran utilidad, es el catéter de Swan-Ganz, que permite medir el gasto cardiaco, las presiones en el circuito pulmonar y el contenido de 02 de la sangre venosa mixta.
4. Insuficiencia mitral aguda. Puede deberse a disfunción o ruptura del músculo papilar. La incidencia de la ruptura de un músculo papilar y/o de sus cuerdas tendinosas en el infarto agudo es del 1%. Ocurre con mas frecuencia en los infartos inferiores, y se caracteriza clínicamente por un soplo pansistolico de regurgitación mitral irradia-do a la axila, dorso y borde izquierdo del esternón. El cuadro clínico puede variar desde la insuficien¬cia cardiaca izquierda leve hasta el edema agudo de pulmón.
El cateterismo derecho con Swan-Ganz muestra la existencia de ondas "V" prominentes en el monitoreo de la presión venocapilar pulmonar, por trasmisión retrograda. El contenido de oxigeno de la aurícula y ventrículo derechos es igual, lo cual hace el diagnostico diferencial con la comunicación interventricular. En este sentido, el ecocardiograma bidimensional y, mejor aun, el eco-Doppler, son de gran utilidad para aclarar los cuadros dudosos. Aun cuando el ecocardiograma bidi¬mensional pueda a veces no revelar una ruptura septal o de un músculo papilar, el Doppler puede demostrar la anormalidad resultante del flujo. Se debe señalar que la sobrevida media en los pacien¬tes con ruptura parcial de un músculo papilar es de tres dias, de manera que se requiere un diagnóstico inmediato con miras a la corrección quirúrgica.
5. Comunicación interventricular (CIV) en el IAM. La ruptura en el tabique interventricular ocurre en menos del 1% de los pacientes con IAM anteroseptal o posteroseptal. La mayoría de los casos tienen lugar en la primera semana del episodio agudo. El tamaño del defecto, y la presencia o ausencia de un aneurisma septal determinan las consecuencias hemodinámicas. Es una complicación que cursa con alia mortalidad: mas del 25% de los casos fallece dentro de los tres primeros dias, el 65% en los primeros quince dias y el 90% dentro de los dos primeros meses.
Los hallazgos clínicos incluyen un froito sistólico palpable y un soplo holosisto1ico rudo en el cuarto o quinto espacio intercostal izquierdo, con irradiación al borde esternal izquierdo, ápex y axila. La insuficiencia cardiaca es global. A veces la diferencia auscultatoria entre un soplo de una CIV y el de una insuficiencia mitral aguda puede ser dificultosa. Se debe colocar un catéter de Swan-Ganz y extraer muestras de sangre de la vena cava superior, la aurícula derecha, el ventrículo derecho y la arteria pulmonar, para determinar el contenido de oxigeno de la misma. Si la diferencia entre la aurícula y el ventrículo derechos es de 1 o mas volúmenes, el diagnóstico de CIV esta hecho, pues se demuestra un cortocircuito de izquierda a derecha. Tambien se puede recurrir al ecocardiograma bidimensional para detectar y localizar el defecto septal posinfarto. Si la CIV es pequeña puede no ser visualizada por eco, pero si mediante eco-Doppler. Si el ecocardiograma bidimensional no encuentra una CIV y el Doppler no detecta flujo turbulento en la region del tabique interventricular, la causa del soplo (y de los síntomas) ser£ casi invariablemente una insuficiencia mitral aguda.
6. Ruptura cardiaca externa. Esta complicación es responsable de alrededor del 10% de los casos fatales intrahospitalarios. Es cuatro veces mas comun en las mujeres, especialmente en pacientes mayores de 60 años que son hipertensos. Ocurre generalmente en la primera semana (tercero al quinto dia) y se observa principalmente en infartos anteriores trasmurales que toman al menos un 10% de la circunferencia ventricular izquierda. Clínicamente se caracteriza por la reaparición brusca del dolor precordial, colapso hemodinámico abrupto, hipotensi6n con signos claros de taponamiento cardiaco, y disociación electromecánica. En esta situación, mientras se inician las maniobras de reanimaci6n, se debe intentar la pericardiocen¬tesis. Si se logra realizar un ecocardiograma apenas se inicia la resucitación, se puede confirmar rápidamente la presencia o ausencia de un hemopericardio. Son cuadros de extrema gravedad.
7.Pericarditis. La pericarditis epistenocardica es muy comun (15% de los casos), presentándose entre el primero y el segundo dia, y desapareciendo a la semana. Se caracteriza por dolor precordial persistente que se puede intensificar con los movimientos respiratorios o los cambios de decúbito, con irradiación a los hombros y dorso. La presen¬cia de un frote sistodiasto1ico confirma el diagnostico.
La aparición de un frote pleural es comun y la existencia de episodios de pericarditis recurrente asociada a neumonitis, puede ser la expresi6n del síndrome autoinmunitario de Dressier, o síndrome posinfarto.
8. Angina posinfarto. Aproximadamente del 10 al 15% de los pacientes con infarto agudo van a desarrollar una angina inestable dentro de la pri¬mera semana del cuadro. Su pronostico es severo, pues se trata de una complicación que se asocia con una elevada incidencia de infarto recurrente o deceso durante la hospitalización. Además, la mortalidad de estos pacientes es alta a los 3 a 6 meses del cuadro.
Este cuadro de angina recurrente, o angina precoz posinfarto, puede deberse a: 1) aumento transitorio de la demanda de oxigeno, por taquiarritmias, anemia, drogas, alteraciones en la volemia, o por problemas cardiacos mecánicos, tales como aneurisma ventricular, insuficiencia mitral, CIV o insuficiencia cardiaca severa; 2) caída transitoria del aporte miocárdico de oxigeno, la cual puede responder a trombosis o agregación plaquetaria transitoria en arterias muy dañadas, o bien a un espasmo arterial coronario próximo al sitio de una estenosis aterosclerosa.
Considerando las múltiples causas posibles, si el paciente tiene angina recurrente se debe investigar por que el angor recurre. Debe practicarse una evaluación clínica completa, exámenes de laboratorio e incluso una angiografía coronaria si se estima que el enfermo se puede beneficiar con terapéuticas invasivas.
Incluso el angor mas tardio, pero que se presenta dentro del mes de ocurrido el I AM y con el paciente aun en reposo, suele responder a causas similares y su pronostico es serio, por lo cual es necesario guiarse por las pautas arriba mencionadas.
9. Infarto del ventrículo derecho (IAM-VD).Si bien el IAM de ventrículo derecho aislado es muy raro, esta afección puede asociarse a un infar¬to trasmural de la pared posteroinferior del ventrículo izquierdo.
El síndrome clínico del IAM-VD aparece entre el 3% y el 8% de los infartos posteroinferiores, si bien se lo encuentra entre el 15 y el 34% de las necropsias y en el 30-70% de los infar¬tos inferiores estudiados con gammagrafia y ecocardiografia. Estas diferencias se deben a que muchas de las lesiones anatomopatológicas no tienen traducción clínica ni hemodinámica. El espectro clínico que origina es muy amplio -puede incluso llevar al shock cardiogenico- pero su diagnostico recae en evidencias clínicas y hemodinámicas de disfunción cardiaca con preponderancia derecha, en el curso de un IAM.Se pueden asi utilizar los siguientes criterios: 1) hallazgos electrocardiográficos de infarto agudo posteroinferior, corroborado por elevación enzimática; 2) evidencia clínica de insuficiencia cardi¬aca derecha dominante, demostrada por ingurgitación yugular, hipertensión venosa central superior a 18 cm H20, y 3) confirmación hemodinámica con catéter de Swan-Ganz de elevación de la presión de fin de diástole ventricular derecha o valores iguales o superiores a la presión capilar pulmonar. Se acompaña además de trastornos de la conducción AV en un alto porcentaje de los casos, que en algunas series llega al 100%.
La presencia de insuficiencia derecha en el curso de un IAM puede responder tambien a insuficien¬cia ventricular izquierda o a un tromboembolismo pulmonar. Corresponde establecer el diagnostico diferencial con estas entidades.
10. Embolia pulmonar. Puede conducir a la muerte súbita, arritmias y/o insuficiencia cardiaca refractaria en el curso de un IAM. No se conoce su frecuencia de aparición. En la mayor parte de los casos el punto de partida son las venas profundas de los miembros inferiores y de la pelvis.
El cuadro clínico dependerá del numero y tamaño de los embolos. Asi, se puede observar desde sudoración, dolor torácico, disnea e hipotensión moderada, hasta insuficiencia cardiaca refractaria al tratamiento, arritmias, shock y muerte. La auscultación solo revelara" un segundo ruido desdoblado y fijo, con P2 acentuado. La arritmia mas frecuente es la fibrilación auricular, y el ECG mostrara desviación del eje a la derecha, rotación horaria, agrandamiento auricular derecho y bloqueo completo de rama derecha. Para confirmar el diagnostico, el estudio mas litil es el centellograma radioisotopico pulmonar. Si este evidencia áreas extensas de hipoperfusion bilaterales, deberá indicarse una arteriografía pulmonar para determinar la necesidad de una embolectomía.
11. Embolias sistémicas. Se trata de una com¬plicaciones rara, y se debe a la formación de trombos murales en la pared del ventrículo izquierdo infartado o de la aurícula, desde donde migran a la periferia. Su pronostico depende del órgano impactado (riñón, cerebro, miembros inferiores, etc.) y del tamaño y jerarquía de la arteria ocluida.